

Robust identification of molecular phenotypes using semi-supervised learning
- PMID: 31138112
- PMCID: PMC6540576
- DOI: 10.1186/s12859-019-2885-3
Robust identification of molecular phenotypes using semi-supervised learning
Abstract
Background: Modern molecular profiling techniques are yielding vast amounts of data from patient samples that could be utilized with machine learning methods to provide important biological insights and improvements in patient outcomes. Unsupervised methods have been successfully used to identify molecularly-defined disease subtypes. However, these approaches do not take advantage of potential additional clinical outcome information. Supervised methods can be implemented when training classes are apparent (e.g., responders or non-responders to treatment). However, training classes can be difficult to define when assessing relative benefit of one therapy over another using gold standard clinical endpoints, since it is often not clear how much benefit each individual patient receives.
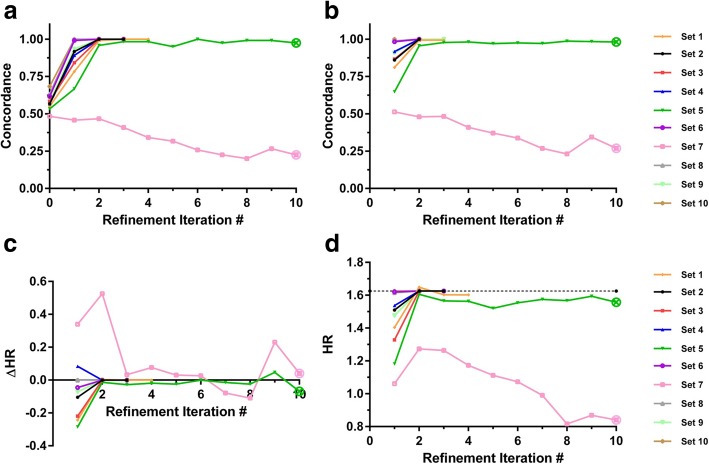
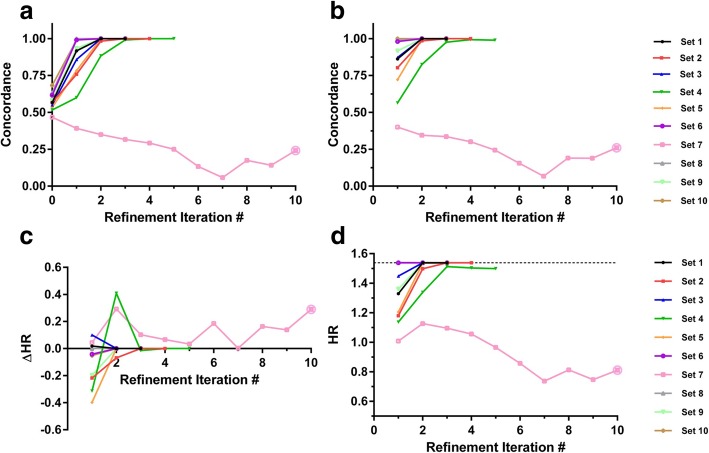
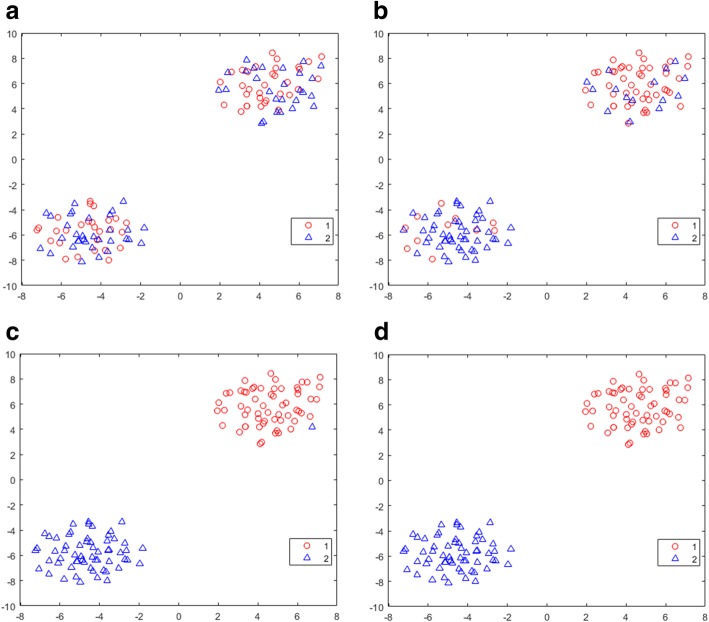
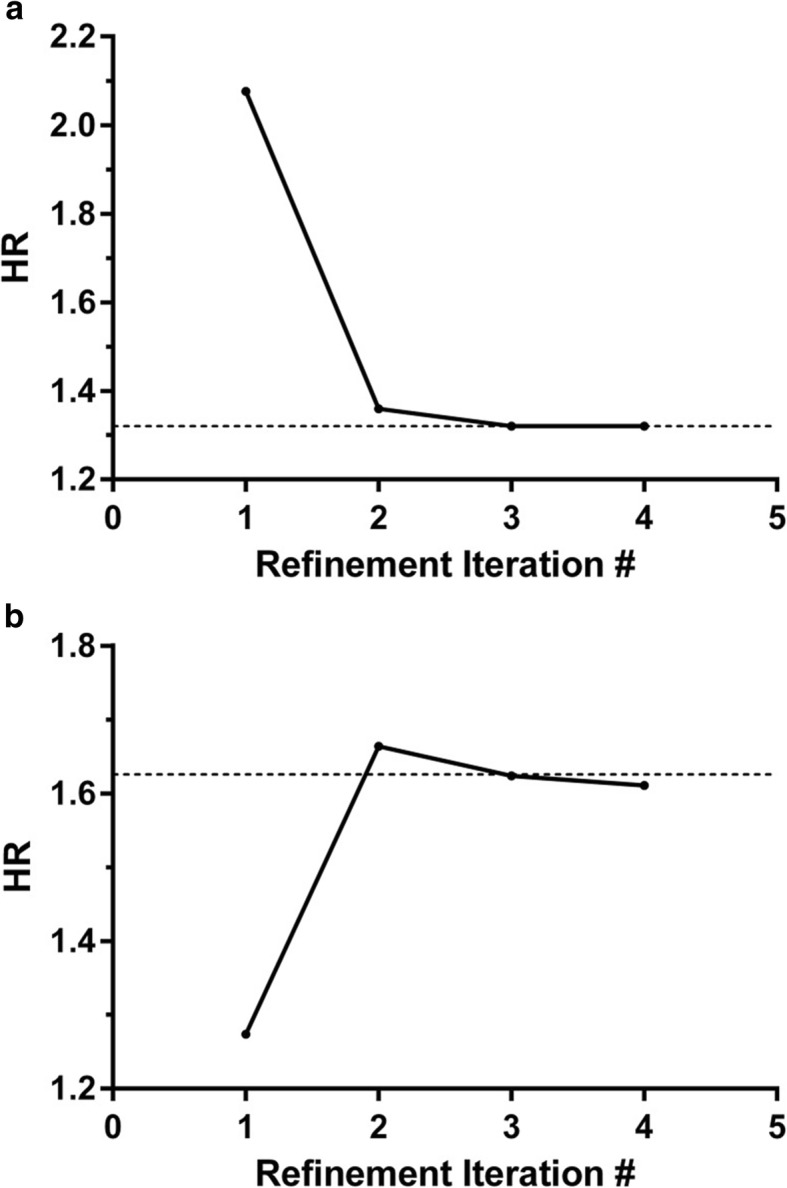
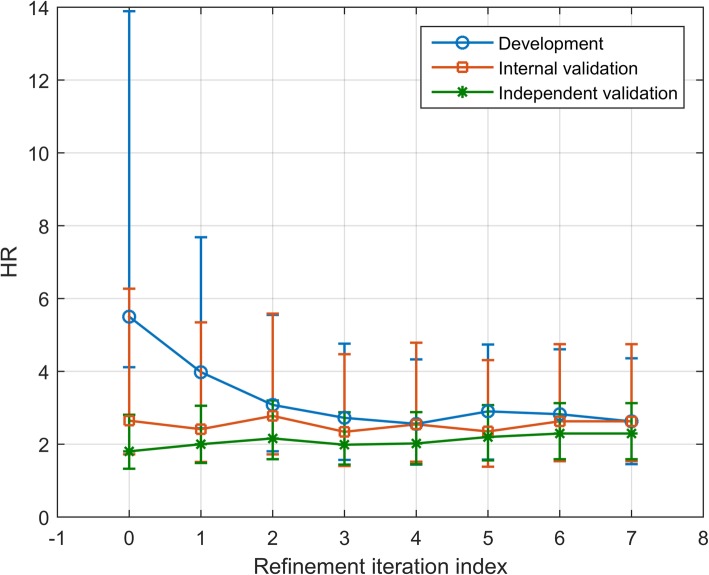
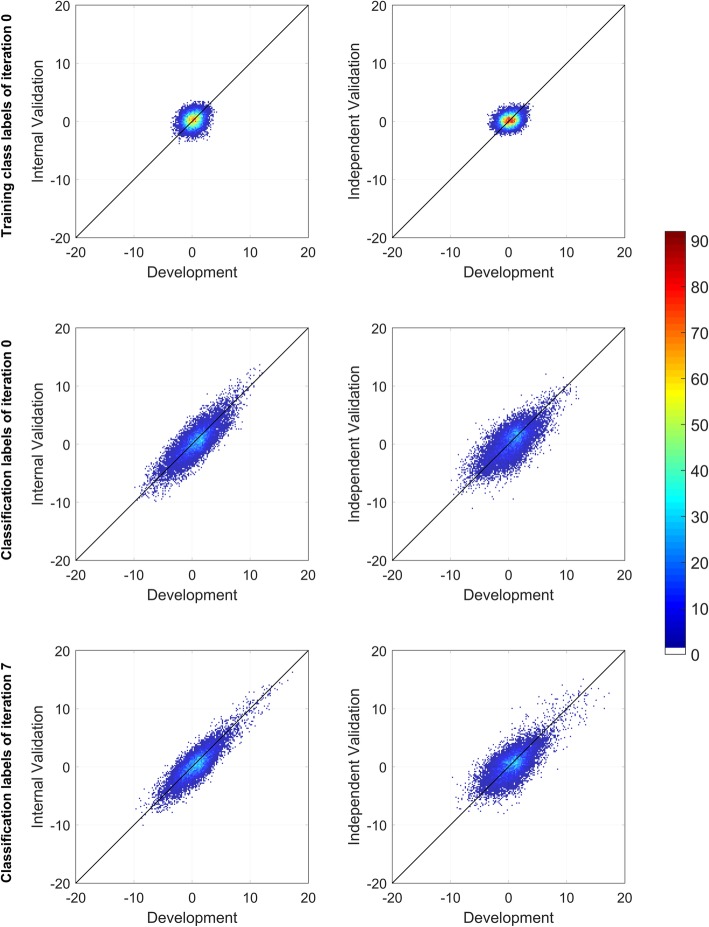
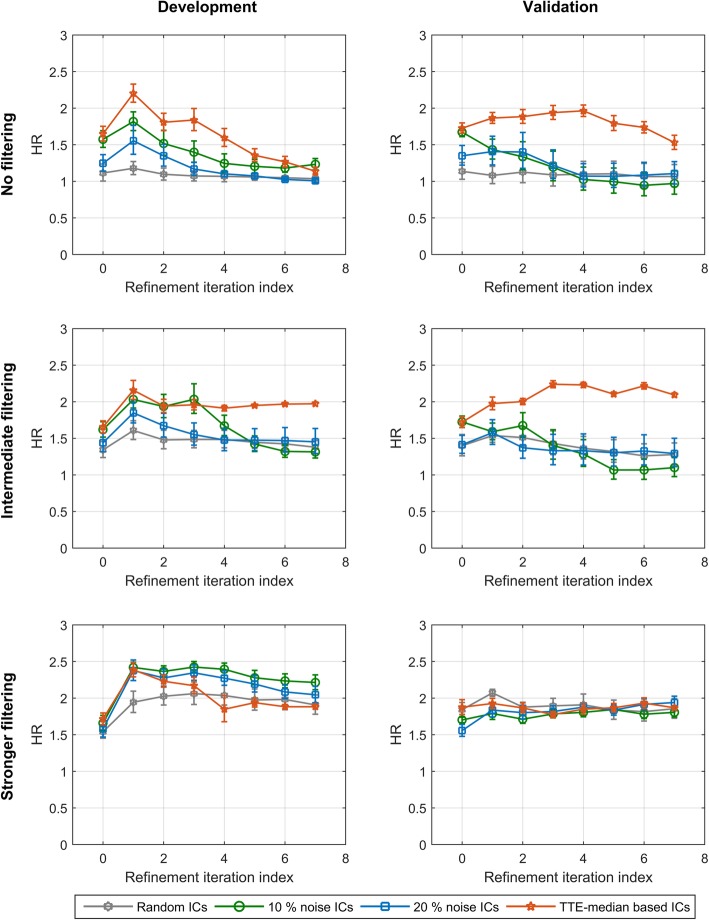
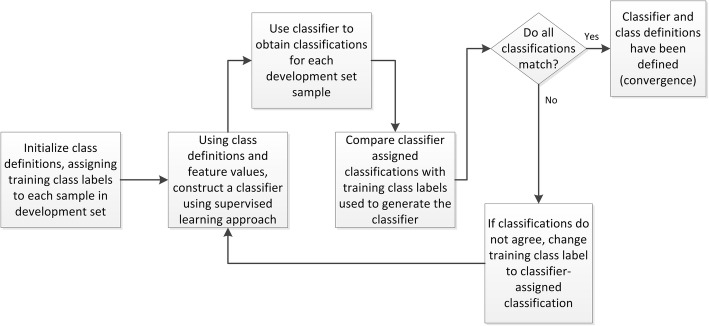
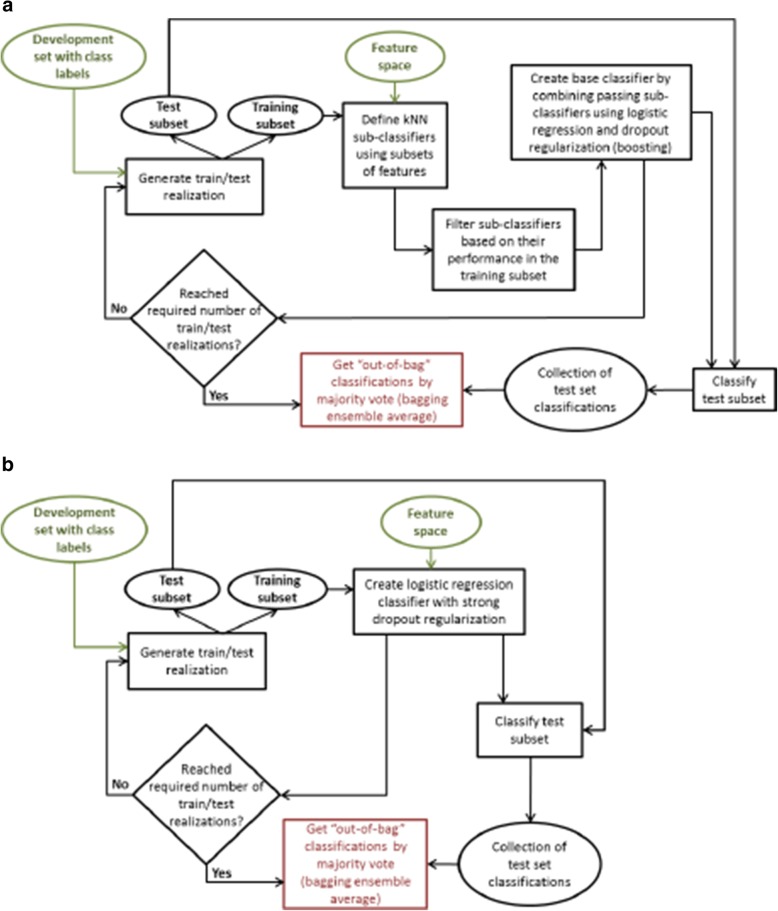
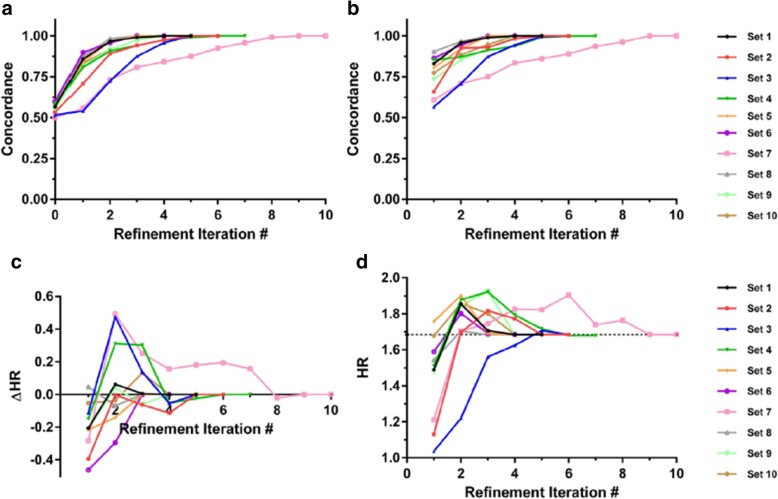
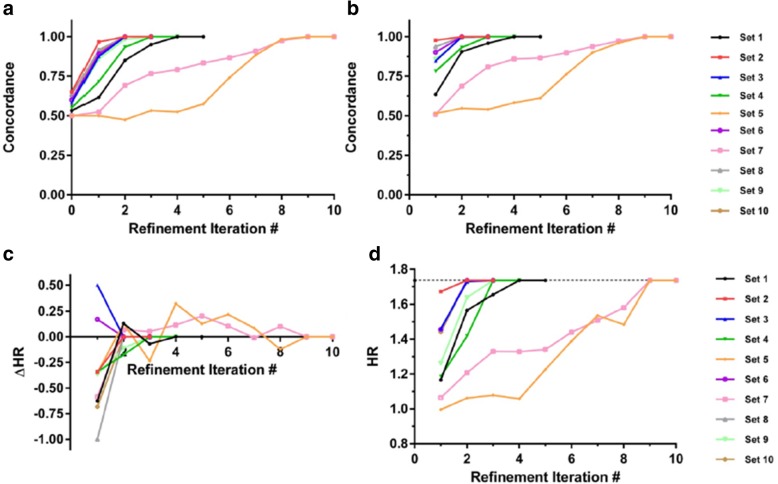
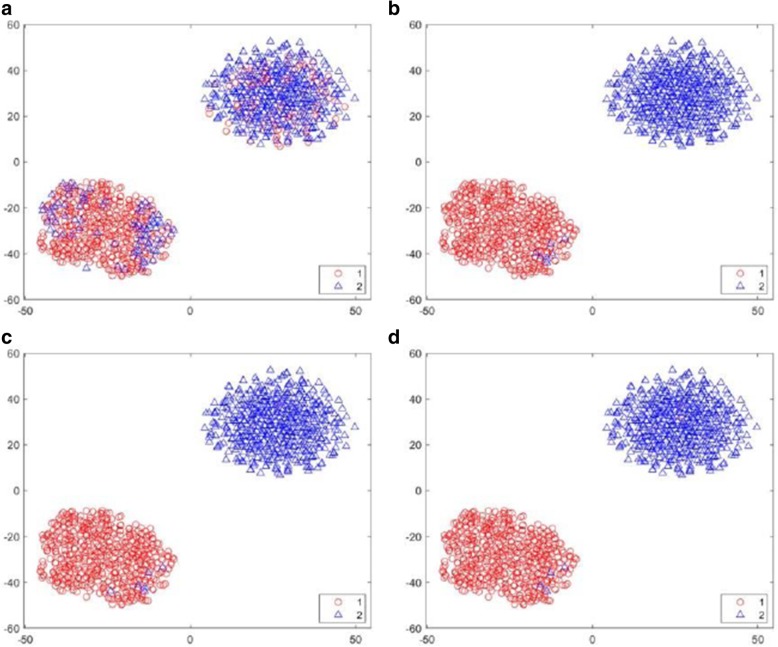
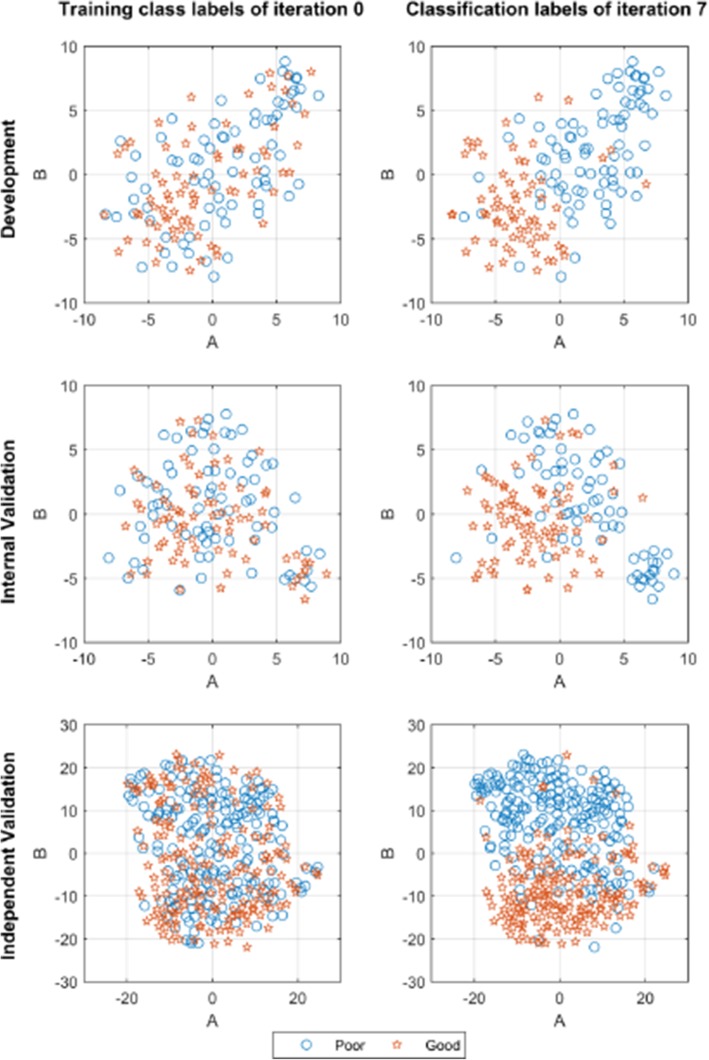
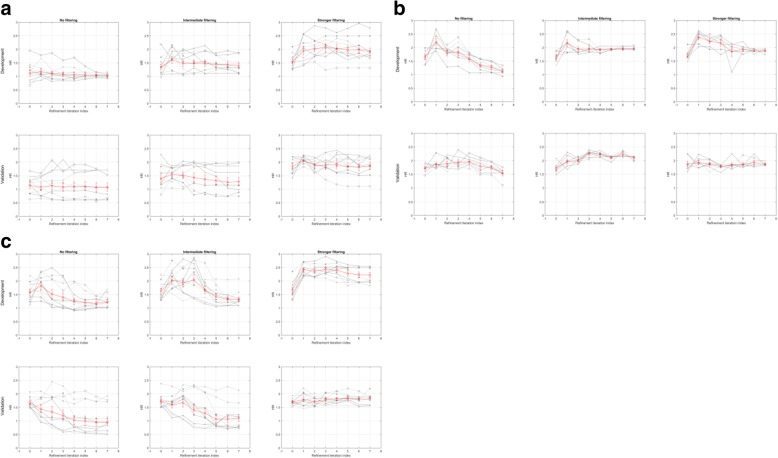
Results: We introduce an iterative approach to binary classification tasks based on the simultaneous refinement of training class labels and classifiers towards self-consistency. As training labels are refined during the process, the method is well suited to cases where training class definitions are not obvious or noisy. Clinical data, including time-to-event endpoints, can be incorporated into the approach to enable the iterative refinement to identify molecular phenotypes associated with a particular clinical variable. Using synthetic data, we show how this approach can be used to increase the accuracy of identification of outcome-related phenotypes and their associated molecular attributes. Further, we demonstrate that the advantages of the method persist in real world genomic datasets, allowing the reliable identification of molecular phenotypes and estimation of their association with outcome that generalizes to validation datasets. We show that at convergence of the iterative refinement, there is a consistent incorporation of the molecular data into the classifier yielding the molecular phenotype and that this allows a robust identification of associated attributes and the underlying biological processes.
Conclusions: The consistent incorporation of the structure of the molecular data into the classifier helps to minimize overfitting and facilitates not only good generalization of classification and molecular phenotypes, but also reliable identification of biologically relevant features and elucidation of underlying biological processes.
Keywords: Clustering; Machine learning; Molecular phenotype; Semi-supervised learning.
Conflict of interest statement
JR and HR are inventors on patents describing the DRC classifier development approach and the simultaneous iterative refinement of classifier and class phenotypes assigned to Biodesix, Inc. All authors are current or former employees of and have or had stock options in Biodesix, Inc.
Figures
References
-
- Sotiriou C, Neo S, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A. 2003;100(18):10393–10398. doi: 10.1073/pnas.1732912100. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
