

Clinical courses and complications of young adults with Autosomal Recessive Polycystic Kidney Disease (ARPKD)
- PMID: 31138820
- PMCID: PMC6538621
- DOI: 10.1038/s41598-019-43488-w
Clinical courses and complications of young adults with Autosomal Recessive Polycystic Kidney Disease (ARPKD)
Abstract
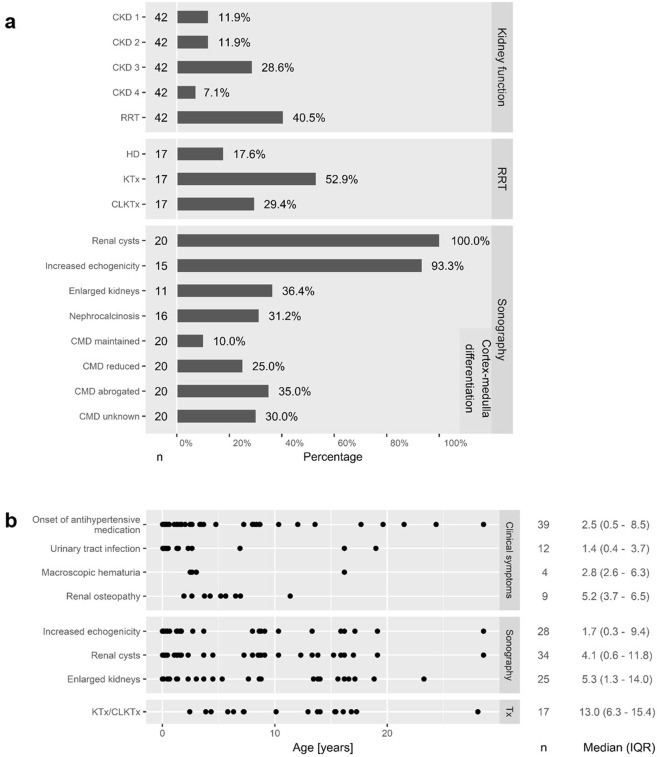
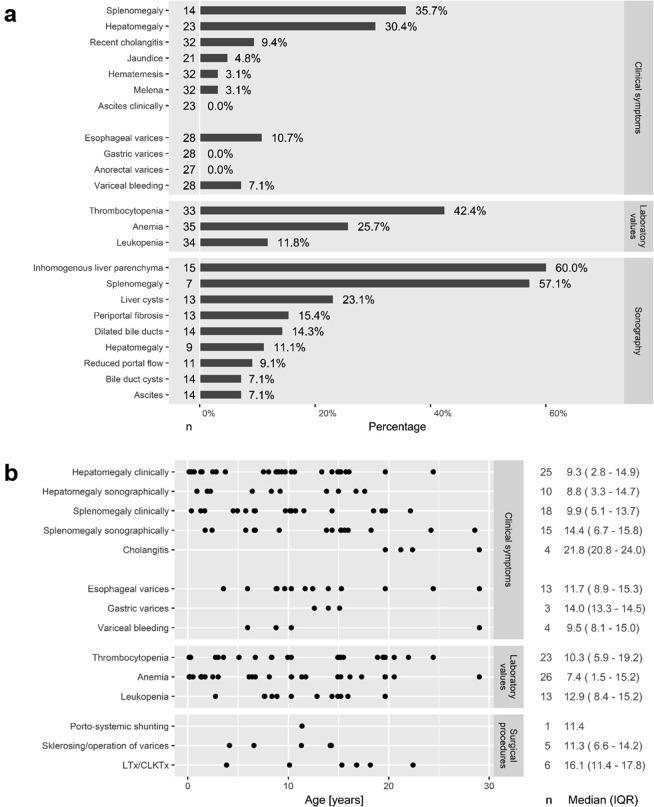
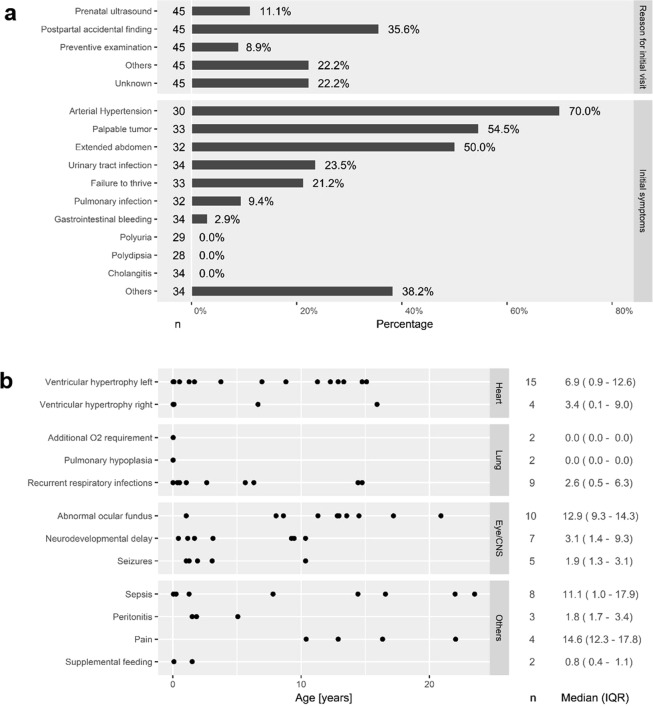
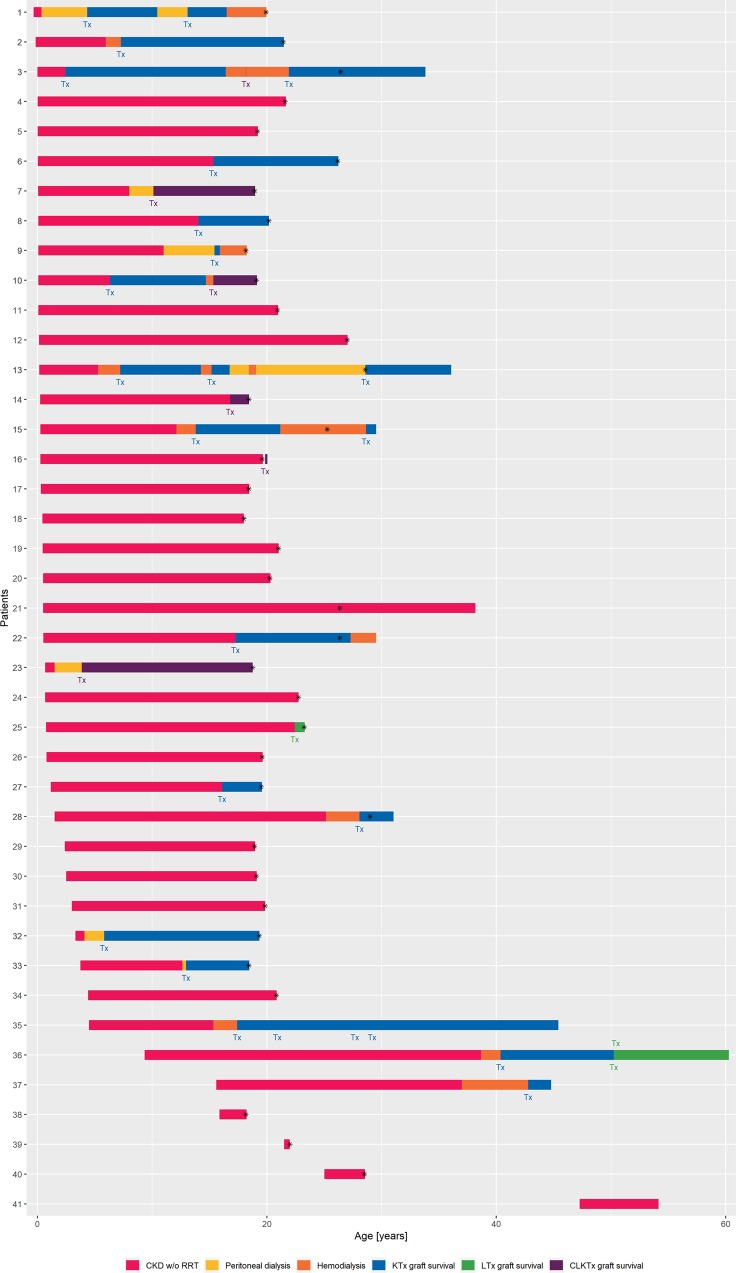
Autosomal recessive polycystic kidney disease (ARPKD) is a severe pediatric hepatorenal disorder with pronounced phenotypic variability. A substantial number of patients with early diagnosis reaches adulthood and some patients are not diagnosed until adulthood. Yet, clinical knowledge about adult ARPKD patients is scarce. Here, we describe forty-nine patients with longitudinal follow-up into young adulthood that were identified in the international ARPKD cohort study ARegPKD. Forty-five patients were evaluated in a cross-sectional analysis at a mean age of 21.4 (±3.3) years describing hepatorenal findings. Renal function of native kidneys was within CKD stages 1 to 3 in more than 50% of the patients. Symptoms of hepatic involvement were frequently detected. Fourteen (31%) patients had undergone kidney transplantation and six patients (13%) had undergone liver transplantation or combined liver and kidney transplantation prior to the visit revealing a wide variability of clinical courses. Hepatorenal involvement and preceding complications in other organs were also evaluated in a time-to-event analysis. In summary, we characterize the broad clinical spectrum of young adult ARPKD patients. Importantly, many patients have a stable renal and hepatic situation in young adulthood. ARPKD should also be considered as a differential diagnosis in young adults with fibrocystic hepatorenal disease.
Conflict of interest statement
M.L. has received honoraria for scientific lectures from Pfizer. Representing the University Hospital of Cologne M.L. has been counselling Otsuka in an advisory board. D.M., represented by KU Leuven University, received an educational grant from Otsuka and participated in an advisory board. K.B., S.K., B.B., T.B., H.B., A.B., M.G., F.G., G.K., L.M., G.M., B.R., K.S., S.S., R.Sh., R.St., S.V., L.W., D.W., E.W., J.D. and F.S. declare no potential conflict of interest.
Figures
References
-
- Lu Hao, Galeano Maria C Rondón, Ott Elisabeth, Kaeslin Geraldine, Kausalya P Jaya, Kramer Carina, Ortiz-Brüchle Nadina, Hilger Nadescha, Metzis Vicki, Hiersche Milan, Tay Shang Yew, Tunningley Robert, Vij Shubha, Courtney Andrew D, Whittle Belinda, Wühl Elke, Vester Udo, Hartleben Björn, Neuber Steffen, Frank Valeska, Little Melissa H, Epting Daniel, Papathanasiou Peter, Perkins Andrew C, Wright Graham D, Hunziker Walter, Gee Heon Yung, Otto Edgar A, Zerres Klaus, Hildebrandt Friedhelm, Roy Sudipto, Wicking Carol, Bergmann Carsten. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nature Genetics. 2017;49(7):1025–1034. doi: 10.1038/ng.3871. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
