

doi: 10.1038/s41587-019-0140-0.
The Kipoi repository accelerates community exchange and reuse of predictive models for genomics
Affiliations
- PMID: 31138913
- PMCID: PMC6777348
- DOI: 10.1038/s41587-019-0140-0
Item in Clipboard
The Kipoi repository accelerates community exchange and reuse of predictive models for genomics
Nat Biotechnol.
2019 Jun.
No abstract available
Conflict of interest statement
The authors declare no competing interests.
Figures
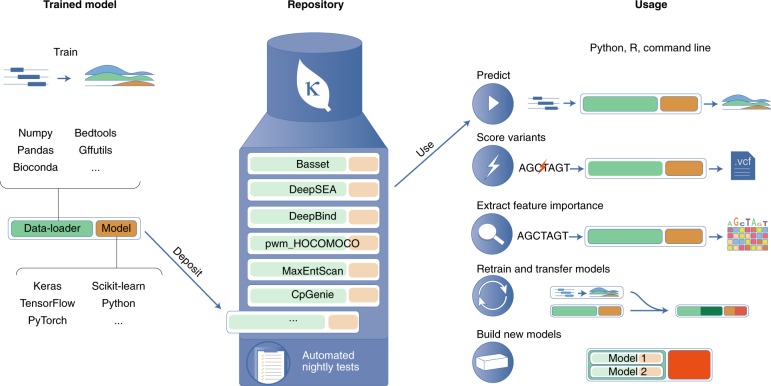
From left to right: at its core, Kipoi defines a programmatic standard for data-loaders and predictive models. Data-loaders translate genomics data into numeric representations that can be used by machine learning models. Kipoi models can be implemented using a broad range of machine-learning frameworks. The Kipoi repository allows users to store and retrieve trained models, together with associated data-loaders. Kipoi models are automatically versioned, nightly tested and systematically documented with examples of their use. Kipoi models can be accessed through unified interfaces from python, R and the command line. All models and their software dependencies can be installed in a fully automatic manner. Kipoi streamlines the application of trained models to make predictions on new data, to score variants stored in the standard variant call format (.vcf) file format, and to assess the effect of variation in the input to model predictions (feature importance score). Moreover, Kipoi models can be adapted to new tasks either by retraining them or by building new composite models that combine existing ones. Newly defined models can be deposited in the repository.
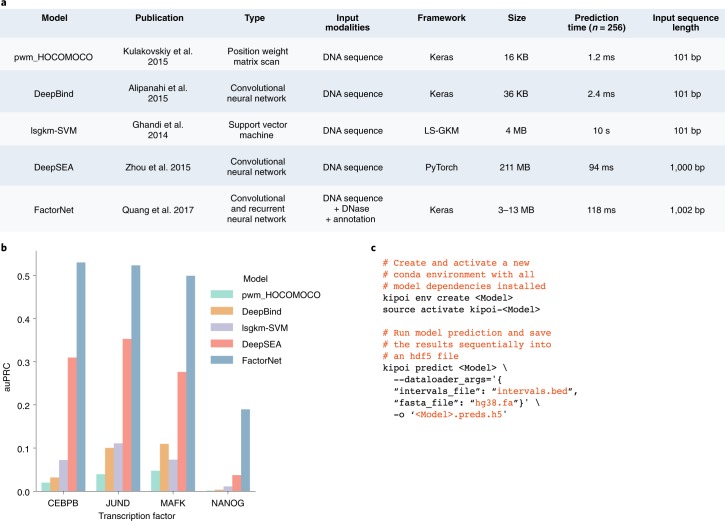
a, Five models for predicting transcription factor binding based on alternative modeling paradigms: first, position weight matrices provided by the HOCOMOCO database; second, lsgkm-SVM, a support vector machine classifier; third, the convolutional neural network DeepBind; fourth, the multi-task convolutional neural network DeepSEA; and finally, FactorNet, a multimodal deep neural network with convolutional and recurrent layers that further integrates chromatin accessibility profile and genomic annotation features. Models differ by both the size of genomic input sequence (DeepSEA and FactorNET consider ~1 kb, whereas other models are based on ~100 bp sequence inputs) and the parametrization complexity, with the total size of stored model parameters ranging from 16 kB (pwm_HOCOMOCO) to 211 MB (DeepSEA). b, Performance of the models in a for predicting ChIP-seq peaks of four transcription factors on held-out data (chromosome 8), quantified using the area under the precision recall curve (auPRC). More complex models yield more accurate predictions than the simpler models such as the commonly used position weight matrices. c, Example use of Kipoi from the command line to install software dependencies, download the model, extract and preprocess the data, and write predictions to a new file. Results as shown in b can be obtained for all Kipoi models listed in a using these generic commands by varying the placeholder <Model>.
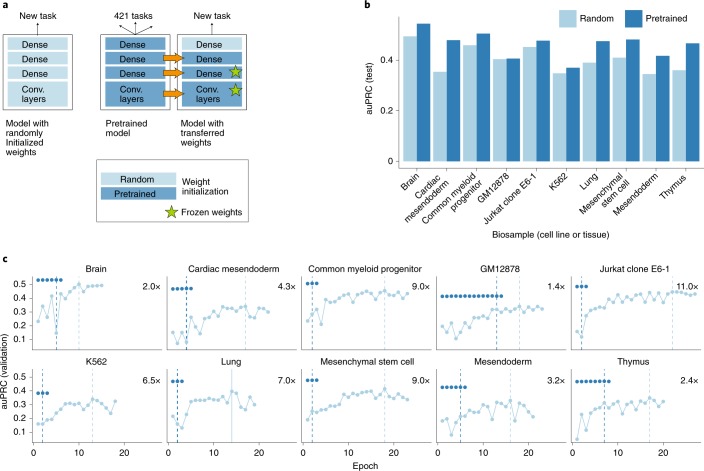
a, Architecture of alternative models for predicting chromatin accessibility from DNA sequence. Model parameters were either randomly initialized (left) or transferred from an existing neural network pretrained on 421 other biosamples (cell lines or tissues, right). b, Predictive performance measured using the area under the precision recall curve (auPRC), comparing randomly initialized (light blue) versus pretrained (dark blue) models. Shown is the performance on held-out data (chromosomes 1, 8 and 21) for 10 biosamples that were not used during pretraining. c, Training curves, showing the auPRC on the validation data (chromosome 9) as a function of the training epoch. The dashed vertical line denotes the training epoch at which the model training was completed. Pretrained models required fewer training epochs than randomly initialized models and achieved more accurate predictions.
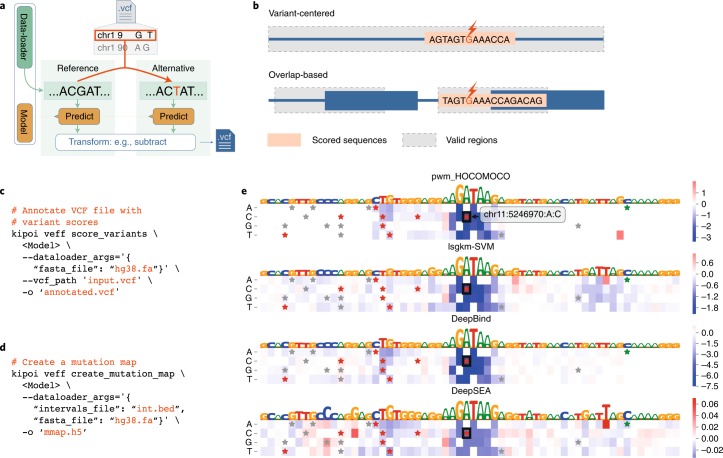
a, Schema of variant effect prediction using in silico mutagenesis. Model predictions calculated for the reference allele and the alternative allele are contrasted and written into an annotated copy of the input variant call format file (.vcf). b, Kipoi uniformly supports variant effect prediction for models that can make predictions anywhere in the genome (top) and also for models that can make predictions only on predefined regions such as exon boundaries (bottom). c, Generic command for variant effect prediction. d, Generic command to compute the importance scores using in silico mutagenesis. e, Feature importance scores visualized as a mutation map (heat map: blue, negative effect; red, positive effect) for variant rs35703285 and the predicted GATA2 binding difference between alleles for four different models. The black boxes in the mutation maps highlight the position and the alternative allele of the respective variant. Stars highlight variants annotated in the human variant database ClinVar, with red indicating likely pathogenic; green, likely benign; gray, uncertain, conflicting significance, and any other type.
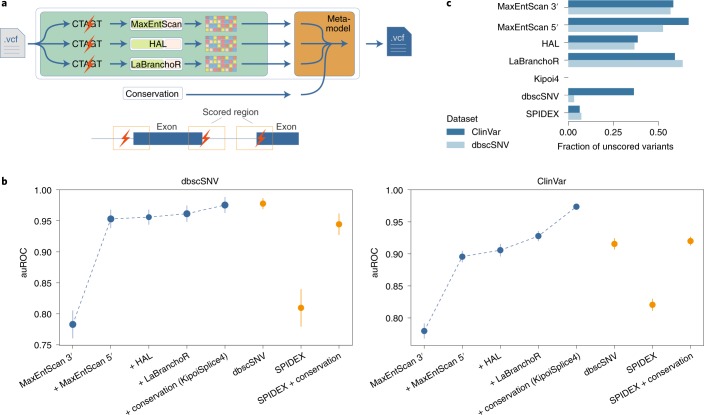
a, Illustration of composite modeling for mRNA splicing. A model trained to distinguish pathogenic from benign splicing region variants is easily constructed by combining Kipoi models for complementary aspects of splicing regulation (MaxEntScan 3′ models the acceptor site, MaxEntScan 5′ and HAL model the donor site, LaBranchoR models the branchpoint) and phylogenetic conservation. These variant scores are combined by logistic regression to predict the variant pathogenicity (orange box). b, Different versions of the ensemble model were trained and evaluated in tenfold cross-validation for the dbscSNV and ClinVar datasets (Supplementary Methods). The four leftmost models are incrementally added to the composite model in chronological order of their publication: the leftmost point only uses information from the MaxEntScan 3′ model, while “+ conservation (KipoiSplice4)” uses all four models and phylogenetic conservation. These performances were compared to a logistic regression model using state-of-the-art splicing variant effect predictors (SPIDEX, SPIDEX + conservation, dbscSNV). KipoiSplice4 achieves state-of-the-art performance on the dbscSNV dataset and outperforms alternative models on ClinVar, which contains a broader range of variants. auROC, area under the receiver operating characteristics curve. c, Fraction of unscored variants for different models in the dbscSNV and ClinVar datasets.
References
Publication types
MeSH terms
Grants and funding
- U01 HG009431/HG/NHGRI NIH HHS/United States
- 1U01HG009431/U.S. Department of Health & Human Services | National Institutes of Health (NIH)/International
- N635290/European Molecular Biology Laboratory (EMBL Heidelberg)/International
- 1DP2OD022870/U.S. Department of Health & Human Services | National Institutes of Health (NIH)/International
- DP2 GM123485/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
