

The Red Fox Y-Chromosome in Comparative Context
- PMID: 31142040
- PMCID: PMC6627929
- DOI: 10.3390/genes10060409
The Red Fox Y-Chromosome in Comparative Context
Abstract
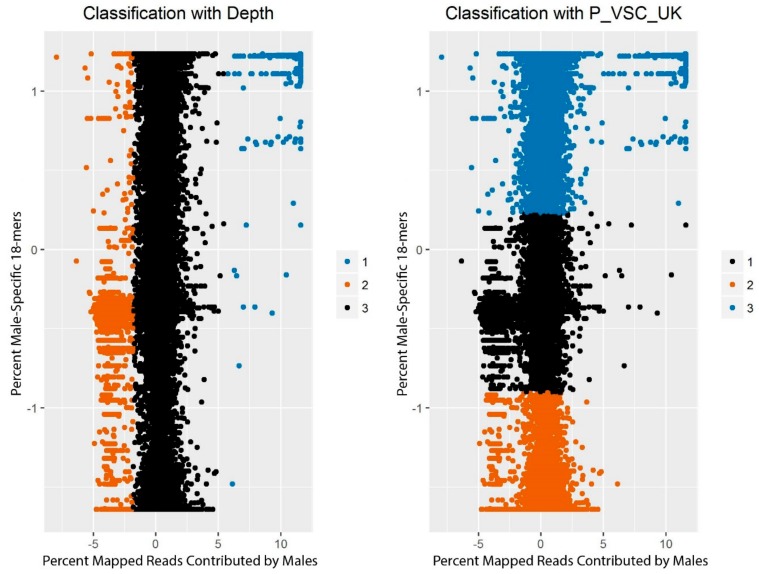
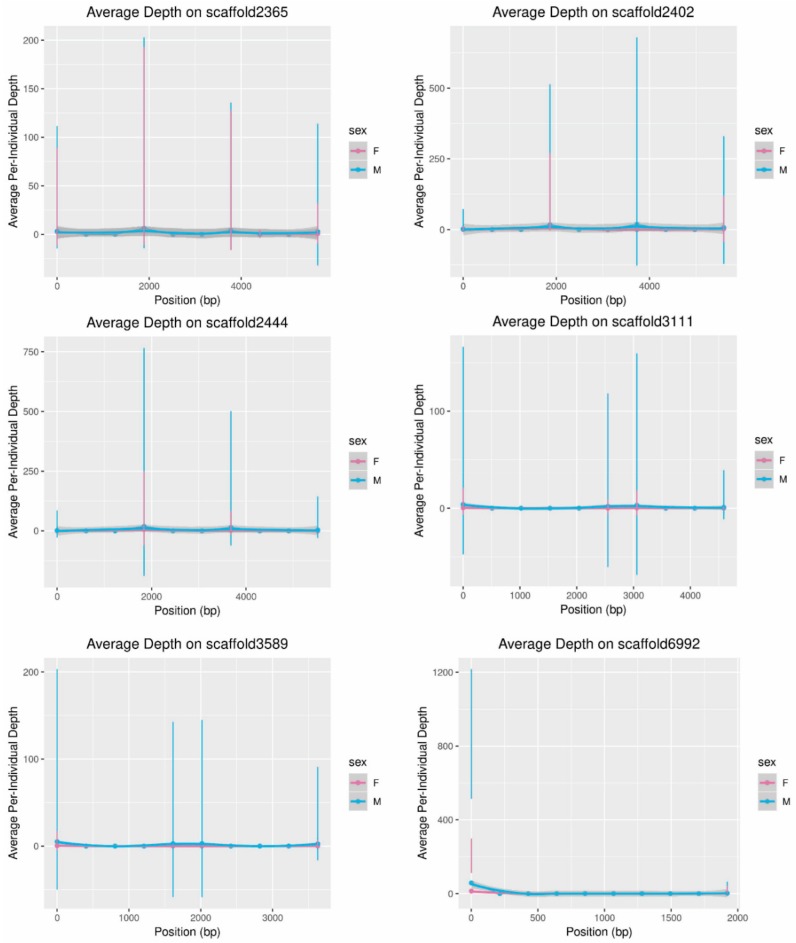
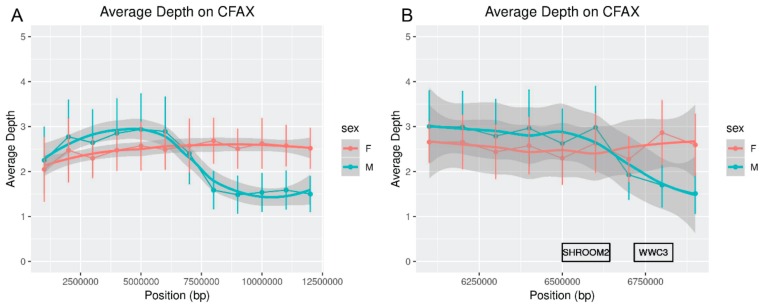
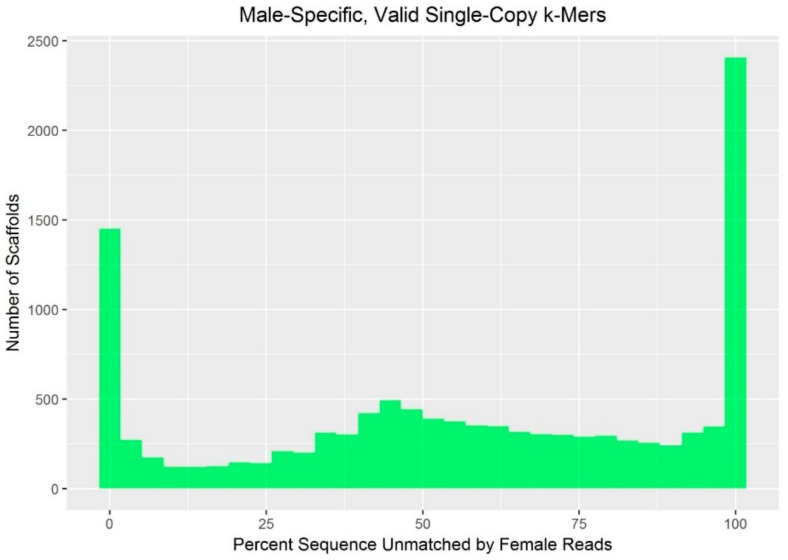
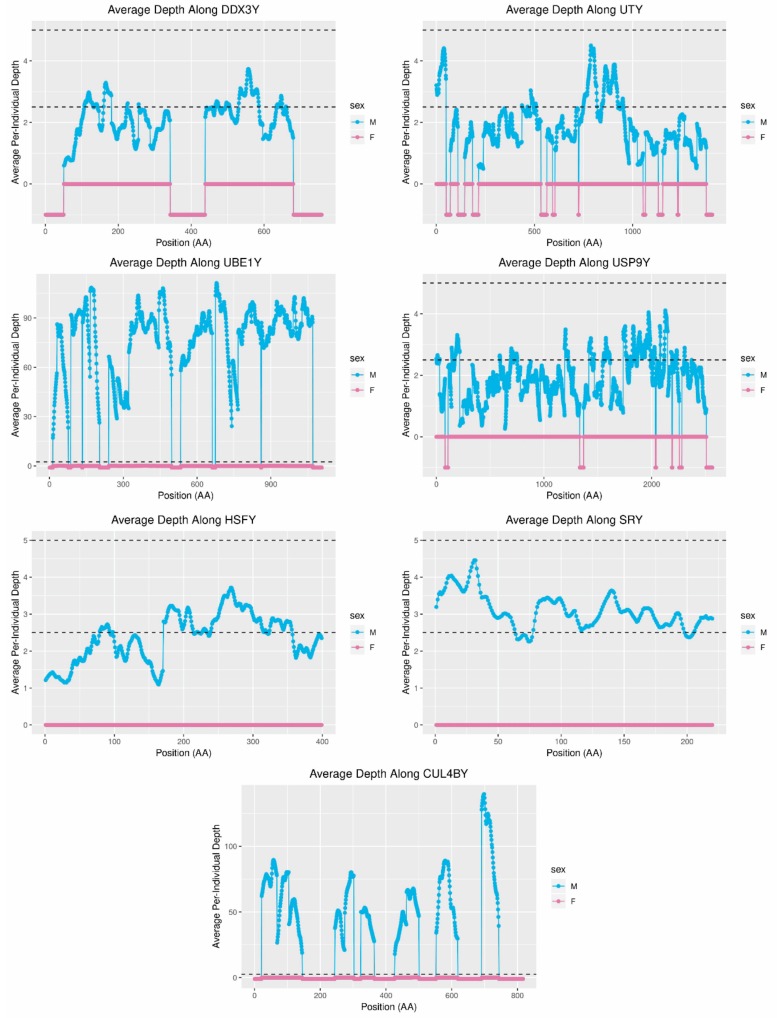
While the number of mammalian genome assemblies has proliferated, Y-chromosome assemblies have lagged behind. This discrepancy is caused by biological features of the Y-chromosome, such as its high repeat content, that present challenges to assembly with short-read, next-generation sequencing technologies. Partial Y-chromosome assemblies have been developed for the cat (Feliscatus), dog (Canislupusfamiliaris), and grey wolf (Canislupuslupus), providing the opportunity to examine the red fox (Vulpesvulpes) Y-chromosome in the context of closely related species. Here we present a data-driven approach to identifying Y-chromosome sequence among the scaffolds that comprise the short-read assembled red fox genome. First, scaffolds containing genes found on the Y-chromosomes of cats, dogs, and wolves were identified. Next, analysis of the resequenced genomes of 15 male and 15 female foxes revealed scaffolds containing male-specific k-mers and patterns of inter-sex copy number variation consistent with the heterogametic chromosome. Analyzing variation across these two metrics revealed 171 scaffolds containing 3.37 Mbp of putative Y-chromosome sequence. The gene content of these scaffolds is consistent overall with that of the Y-chromosome in other carnivore species, though the red fox Y-chromosome carries more copies of BCORY2 and UBE1Y than has been reported in related species and fewer copies of SRY than in other canids. The assignment of these scaffolds to the Y-chromosome serves to further characterize the content of the red fox draft genome while providing resources for future analyses of canid Y-chromosome evolution.
Keywords: BCORY2; MSY; UBE1Y; Vulpes vulpes; Y-chromosome; Y-chromosome genes; carnivore; copy-number variation; next-generation sequencing; sex chromosomes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
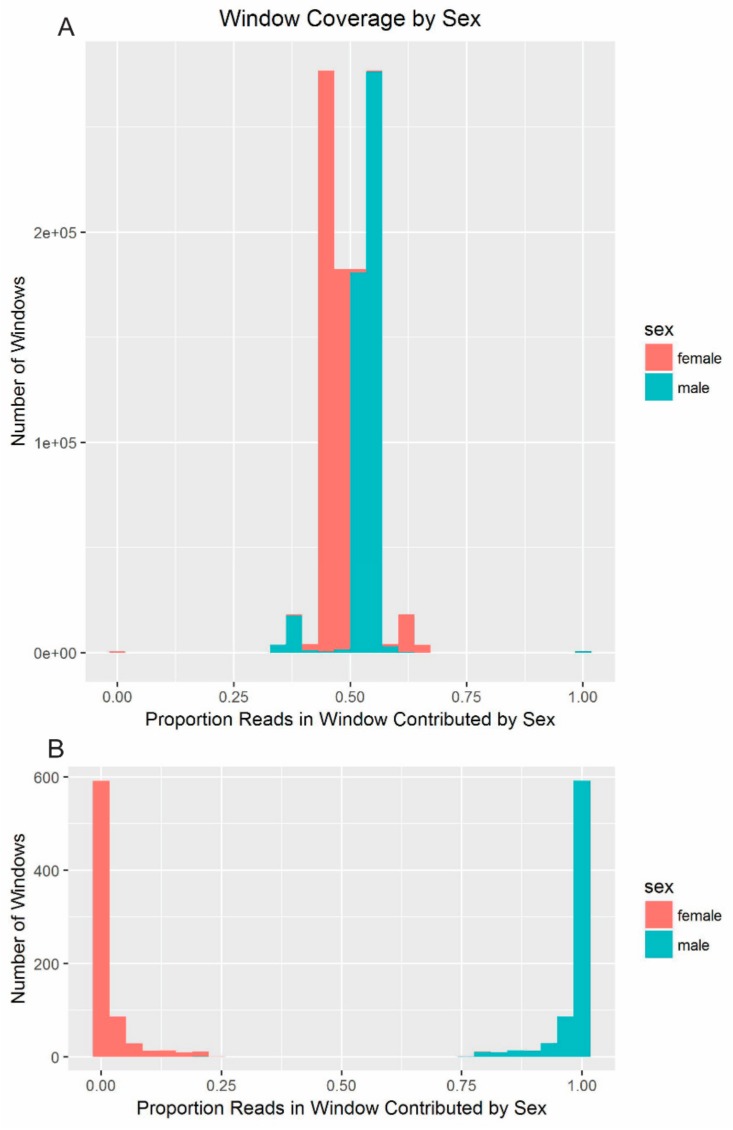
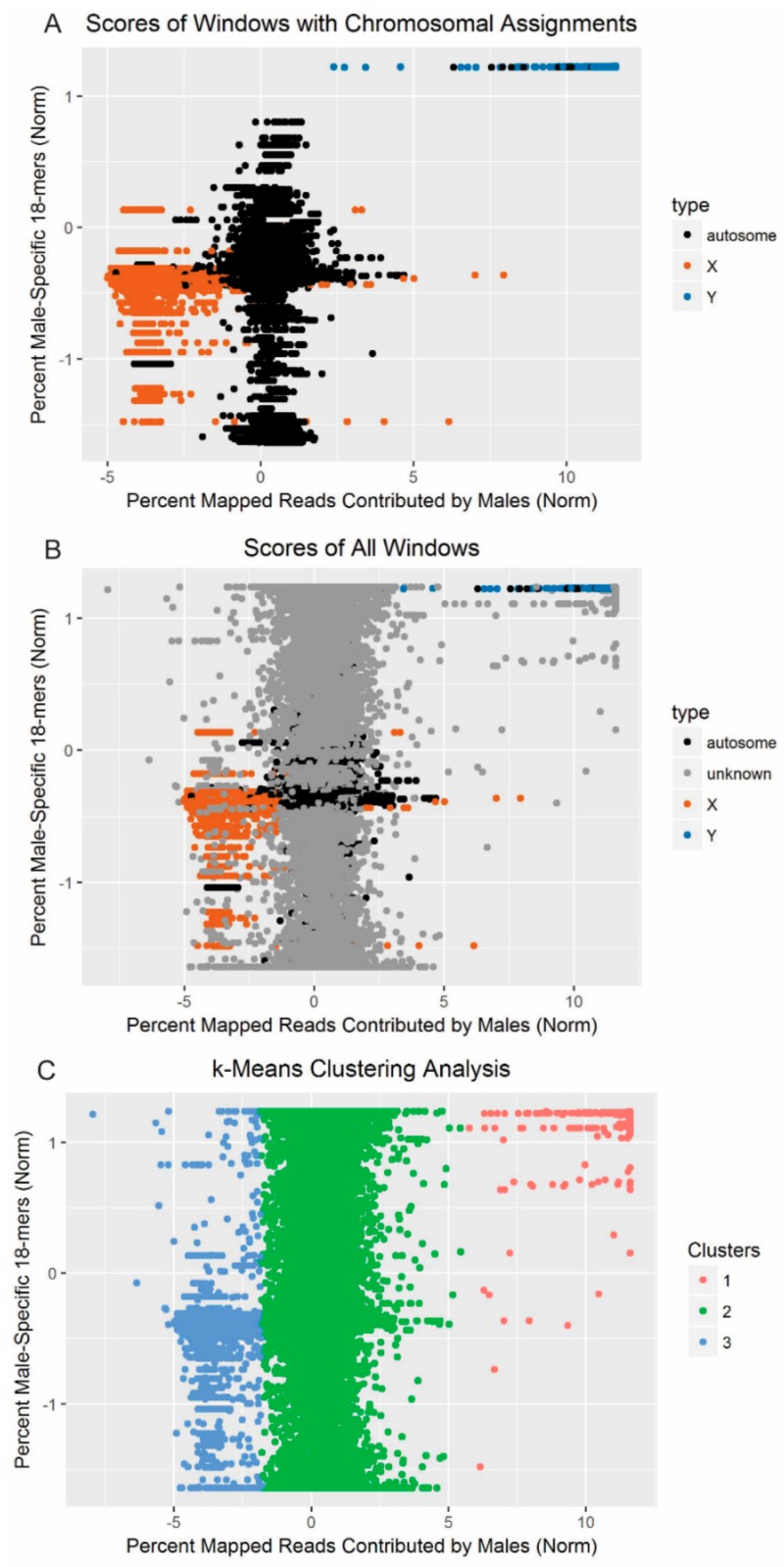
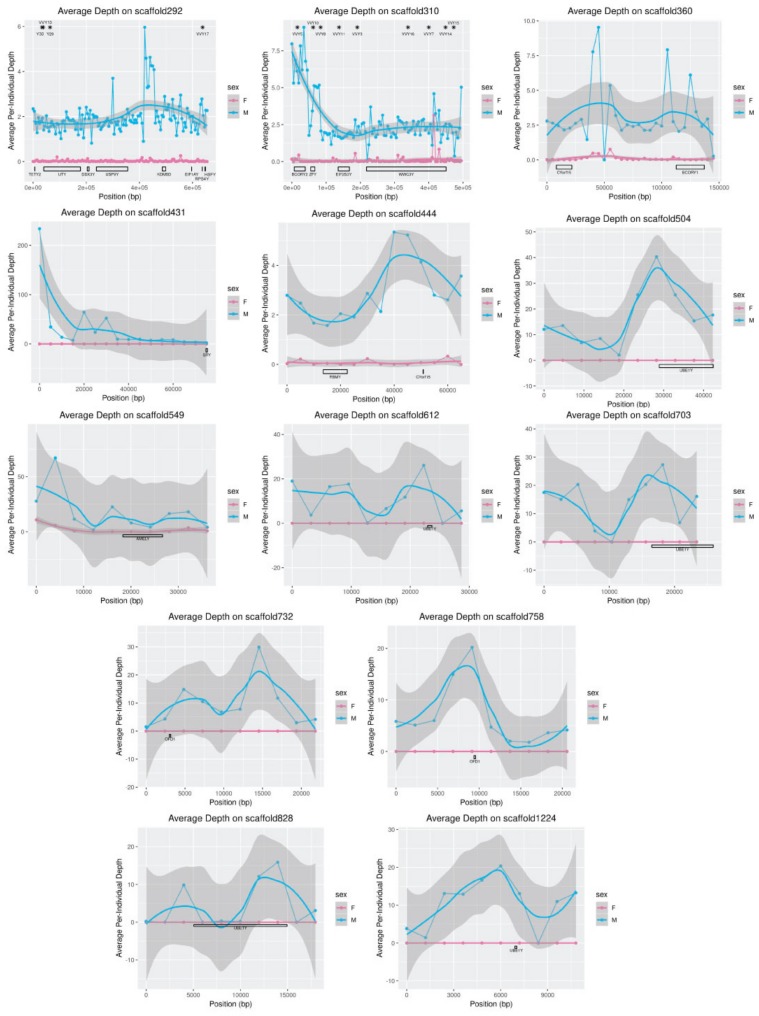
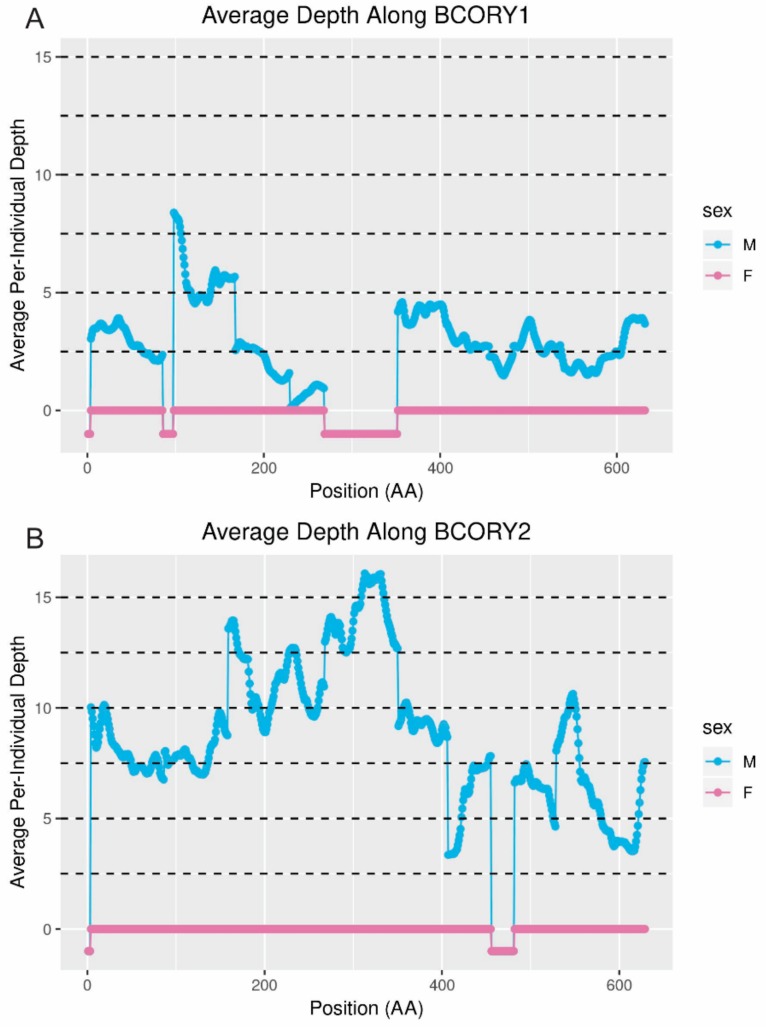
Similar articles
-
Construction of Red Fox Chromosomal Fragments from the Short-Read Genome Assembly.Genes (Basel). 2018 Jun 20;9(6):308. doi: 10.3390/genes9060308. Genes (Basel). 2018. PMID: 29925783 Free PMC article.
-
Y-Chromosome Markers for the Red Fox.J Hered. 2017 Sep 1;108(6):678-685. doi: 10.1093/jhered/esx066. J Hered. 2017. PMID: 28821189 Free PMC article.
-
Synteny search identifies carnivore Y chromosome for evolution of male specific genes.Integr Zool. 2019 May;14(3):224-234. doi: 10.1111/1749-4877.12352. Integr Zool. 2019. PMID: 30019860
-
Y and W Chromosome Assemblies: Approaches and Discoveries.Trends Genet. 2017 Apr;33(4):266-282. doi: 10.1016/j.tig.2017.01.008. Epub 2017 Feb 22. Trends Genet. 2017. PMID: 28236503 Review.
-
The Y chromosomes of the great apes.Hum Genet. 2017 May;136(5):511-528. doi: 10.1007/s00439-017-1769-8. Epub 2017 Mar 6. Hum Genet. 2017. PMID: 28265767 Review.
Cited by
-
Expression Evolution of Ancestral XY Gametologs across All Major Groups of Placental Mammals.Genome Biol Evol. 2020 Nov 3;12(11):2015-2028. doi: 10.1093/gbe/evaa173. Genome Biol Evol. 2020. PMID: 32790864 Free PMC article.
-
Sequencing Red Fox Y Chromosome Fragments to Develop Phylogenetically Informative SNP Markers and Glimpse Male-Specific Trans-Pacific Phylogeography.Genes (Basel). 2021 Jan 14;12(1):97. doi: 10.3390/genes12010097. Genes (Basel). 2021. PMID: 33466657 Free PMC article.
-
An 8.22 Mb Assembly and Annotation of the Alpaca (Vicugna pacos) Y Chromosome.Genes (Basel). 2021 Jan 16;12(1):105. doi: 10.3390/genes12010105. Genes (Basel). 2021. PMID: 33467186 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
