

Predictors of death or lung transplant after a diagnosis of idiopathic pulmonary fibrosis: insights from the IPF-PRO Registry
- PMID: 31142314
- PMCID: PMC6542049
- DOI: 10.1186/s12931-019-1043-9
Predictors of death or lung transplant after a diagnosis of idiopathic pulmonary fibrosis: insights from the IPF-PRO Registry
Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is a progressive disease with a variable clinical course and high mortality. We used data from a large national US registry of patients with IPF to investigate relationships between patient characteristics, including markers of disease severity, and mortality.
Methods: The analysis cohort comprised patients enrolled in the IPF-PRO Registry from its inception on 5 June 2014 to 26 October 2017. The primary criterion for inclusion in this registry is that patients must be diagnosed or confirmed with IPF at the enrolling centre within 6 months. Associations between patient characteristics and markers of disease severity at enrolment and mortality outcomes were investigated using univariable, multivariable and adjustment models.
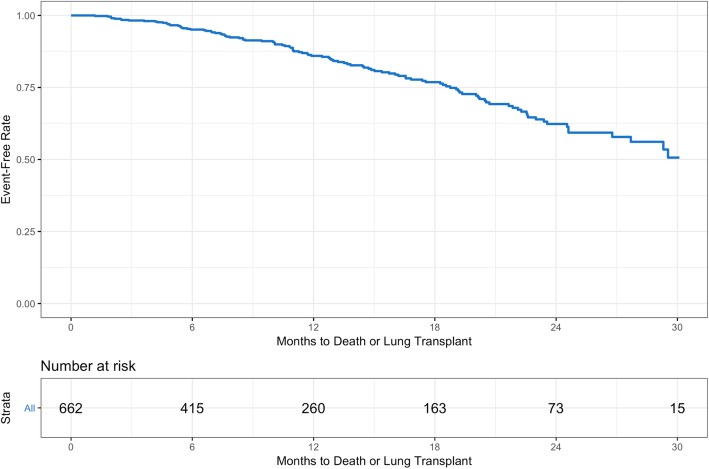
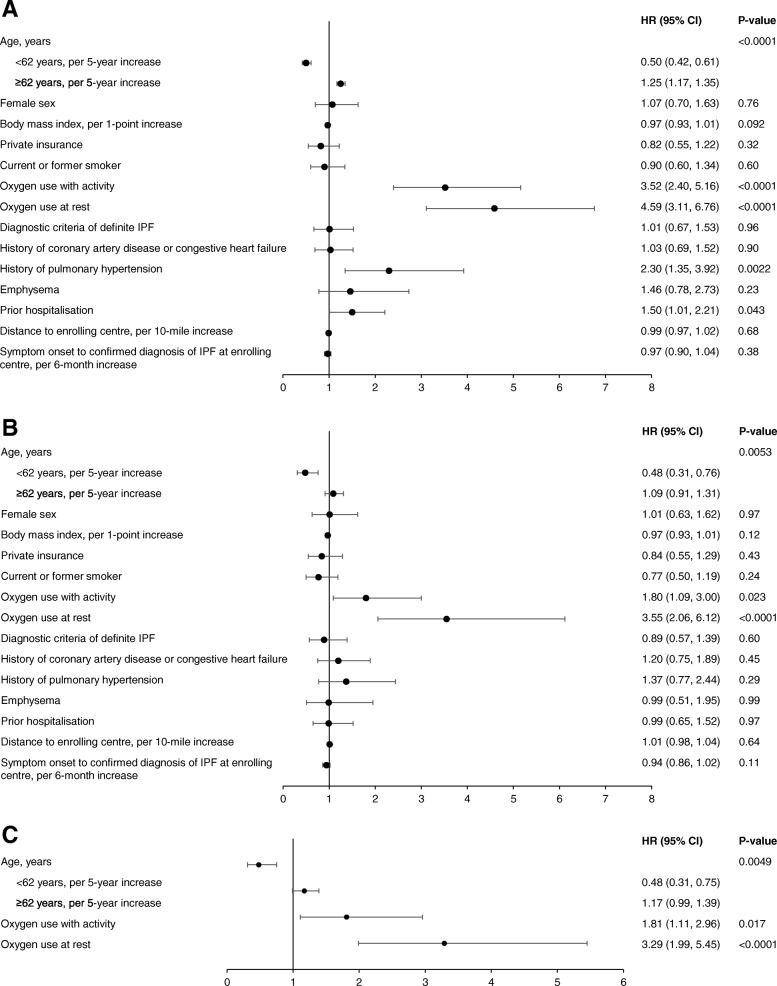
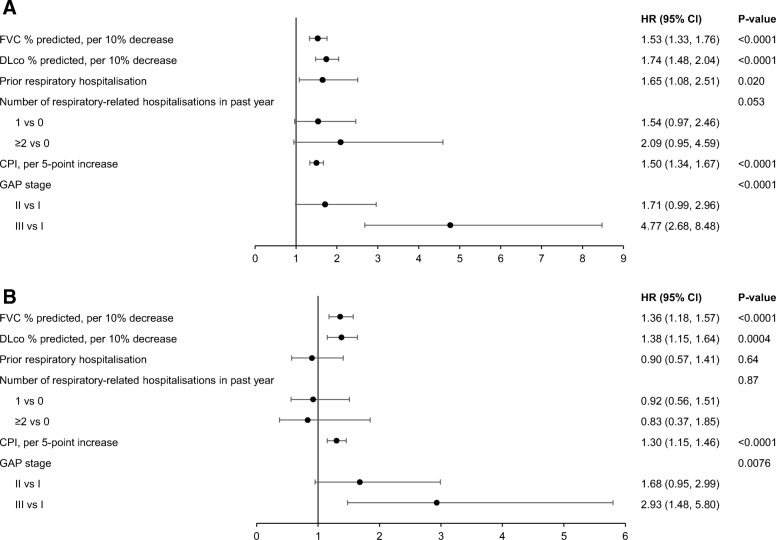
Results: Among 662 patients enrolled, 111 patients died or had a lung transplant over a follow-up period of 30 months. The probability of being free of both events at month 30 was 50.6% (95% CI: 40.0, 60.2). When patient characteristics and markers of disease severity were jointly examined in a multivariable analysis, oxygen use at rest (hazard ratio [HR] 2.44 [95% CI: 1.45, 4.10]), lower forced vital capacity (FVC) % predicted (HR 1.28 [95% CI: 1.10, 1.49] per 10% decrease) and diffusion capacity for carbon monoxide (DLco) % predicted (HR 1.25 [95% CI: 1.04, 1.51] per 10% decrease) were significantly associated with increased risk of death or lung transplant. The risk of death or lung transplant increased with increasing age in patients ≥62 years old (HR 1.18 [95% CI: 0.99, 1.40] per 5-year increase), and decreased with increasing age in patients <62 years old (HR 0.60 [95% CI: 0.39, 0.92] per 5-year increase).
Conclusions: In an observational US registry of patients with IPF, oxygen use at rest, lower FVC % predicted, and lower DLco % predicted were associated with risk of death or lung transplant. An audio podcast of the lead author discussing these data can be downloaded from: http://www.usscicomms.com/respiratory/snyder/IPF-PROsurvival1/ .
Trial registration: ClinicalTrials.gov number: NCT01915511 .
Conflict of interest statement
LS, MLN, ASH, EOB and SP are faculty members in the Duke Clinical Research Institute (DCRI), which receives funding support from Boehringer Ingelheim Pharmaceuticals, Inc. (BIPI) to coordinate the IPF-PRO Registry. CSC, SB and TL are employees of BIPI. MG reports personal fees from the France Foundation, Pulmonary Fibrosis Foundation, Boehringer Ingelheim, and Genentech. DAC reports personal fees from Boehringer Ingelheim, Genentech, and Biogen. JdA reports personal fees from Boehringer Ingelheim. RJK reports grants and personal fees from Boehringer Ingelheim and Genentech; personal fees from MedImmune and Gilead; and grants from Afferent, Bristol-Myers Squibb, the National Institutes of Health. HJK reports an educational grant from Genentech.
Figures


References
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Medical
