

Host-Pathogen Interactions in Gram-Positive Bacterial Pneumonia
- PMID: 31142498
- PMCID: PMC6589866
- DOI: 10.1128/CMR.00107-18
Host-Pathogen Interactions in Gram-Positive Bacterial Pneumonia
Abstract
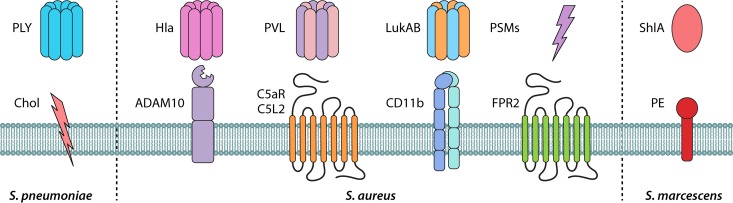
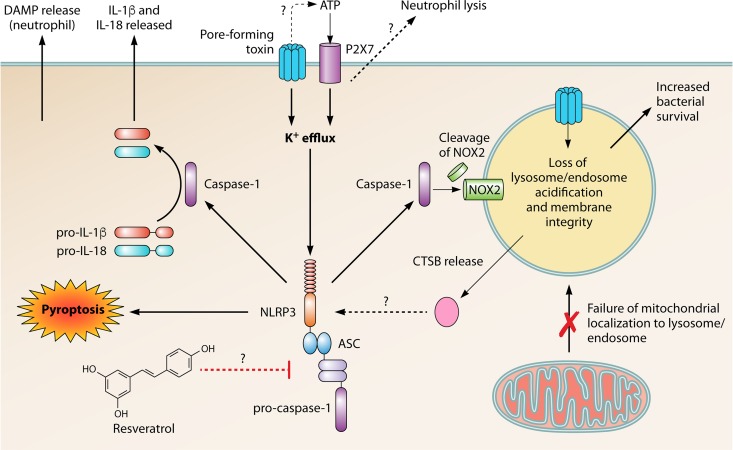
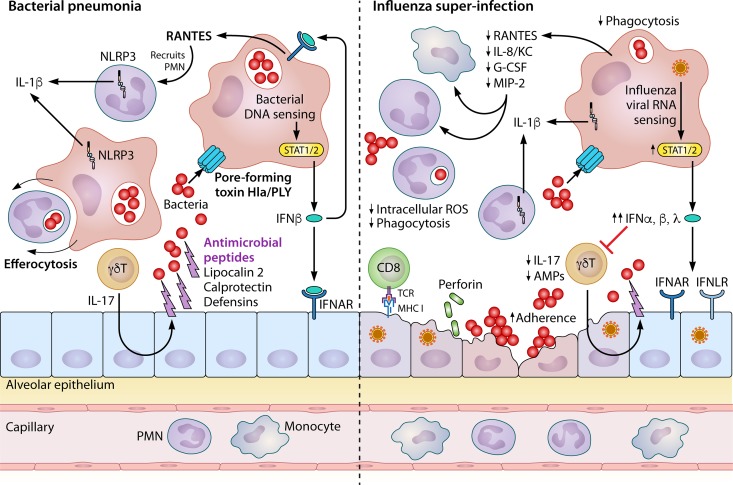
Community-acquired pneumonia (CAP) is a leading cause of morbidity and mortality worldwide. Despite broad literature including basic and translational scientific studies, many gaps in our understanding of host-pathogen interactions remain. In this review, pathogen virulence factors that drive lung infection and injury are discussed in relation to their associated host immune pathways. CAP epidemiology is considered, with a focus on Staphylococcus aureus and Streptococcus pneumoniae as primary pathogens. Bacterial factors involved in nasal colonization and subsequent virulence are illuminated. A particular emphasis is placed on bacterial pore-forming toxins, host cell death, and inflammasome activation. Identified host-pathogen interactions are then examined by linking pathogen factors to aberrant host response pathways in the context of acute lung injury in both primary and secondary infection. While much is known regarding bacterial virulence and host immune responses, CAP management is still limited to mostly supportive care. It is likely that improvements in therapy will be derived from combinatorial targeting of both pathogen virulence factors and host immunomodulation.
Keywords: inflammation; influenza; lung; staphylococcus; streptococcus; superinfection.
Copyright © 2019 American Society for Microbiology.
Figures
References
-
- Jain S, Self WH, Wunderink RG, Fakhran S, Balk R, Bramley AM, Reed C, Grijalva CG, Anderson EJ, Courtney DM, Chappell JD, Qi C, Hart EM, Carroll F, Trabue C, Donnelly HK, Williams DJ, Zhu Y, Arnold SR, Ampofo K, Waterer GW, Levine M, Lindstrom S, Winchell JM, Katz JM, Erdman D, Schneider E, Hicks LA, McCullers JA, Pavia AT, Edwards KM, Finelli L, CDC EPIC Study Team. 2015. Community-acquired pneumonia requiring hospitalization among U.S. adults. N Engl J Med 373:415–427. doi:10.1056/NEJMoa1500245. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
