

Cystic Fibrosis and Pseudomonas aeruginosa: the Host-Microbe Interface
- PMID: 31142499
- PMCID: PMC6589863
- DOI: 10.1128/CMR.00138-18
Cystic Fibrosis and Pseudomonas aeruginosa: the Host-Microbe Interface
Abstract
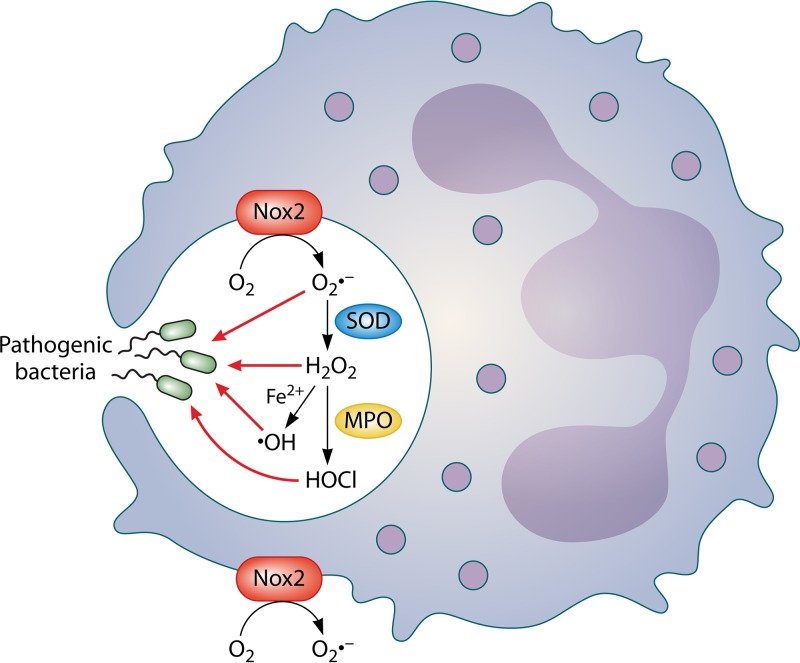
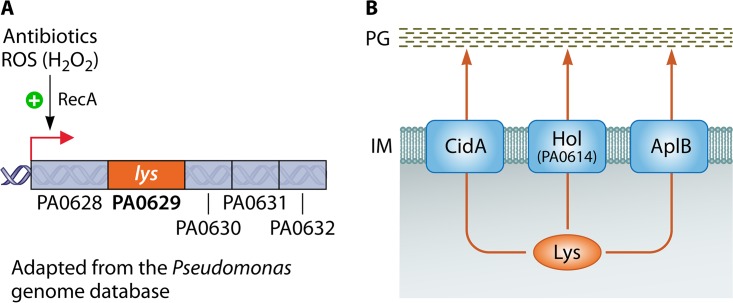
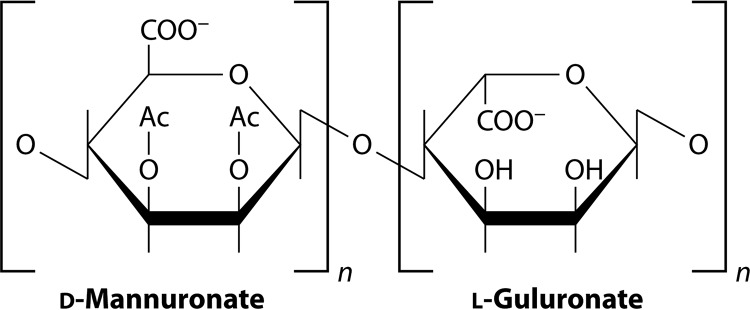
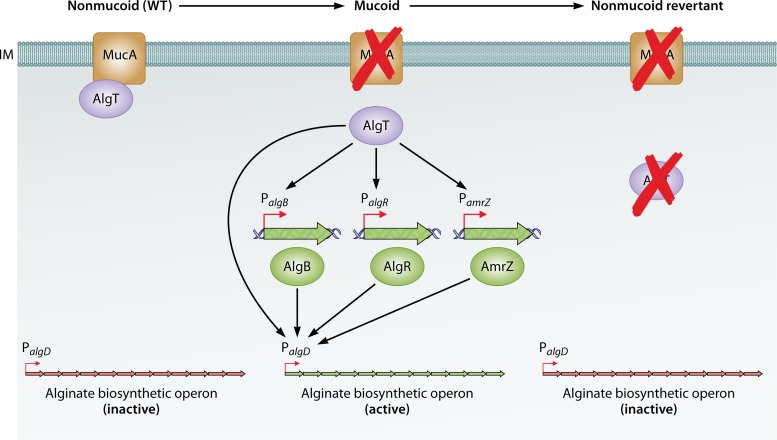
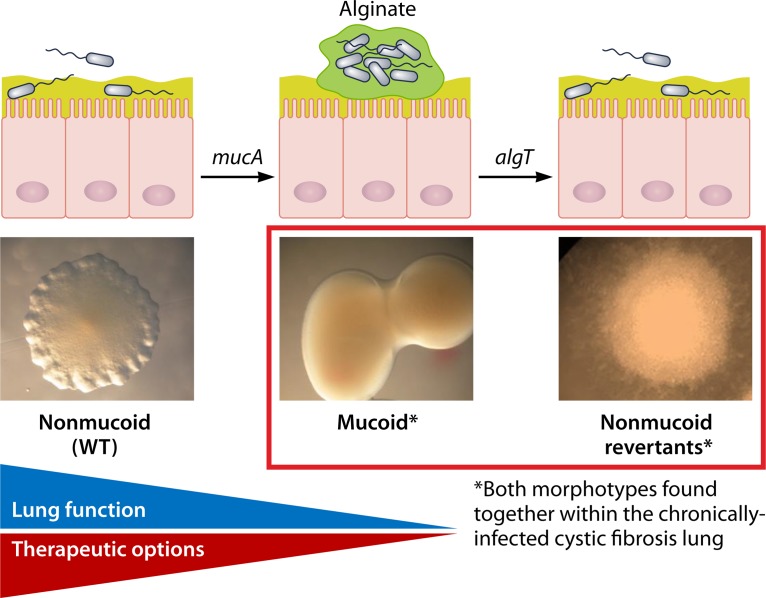
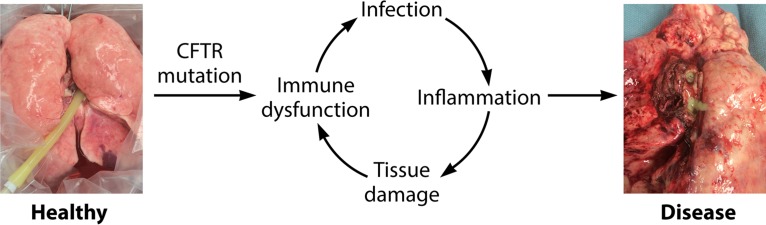
In human pathophysiology, the clash between microbial infection and host immunity contributes to multiple diseases. Cystic fibrosis (CF) is a classical example of this phenomenon, wherein a dysfunctional, hyperinflammatory immune response combined with chronic pulmonary infections wreak havoc upon the airway, leading to a disease course of substantial morbidity and shortened life span. Pseudomonas aeruginosa is an opportunistic pathogen that commonly infects the CF lung, promoting an accelerated decline of pulmonary function. Importantly, P. aeruginosa exhibits significant resistance to innate immune effectors and to antibiotics, in part, by expressing specific virulence factors (e.g., antioxidants and exopolysaccharides) and by acquiring adaptive mutations during chronic infection. In an effort to review our current understanding of the host-pathogen interface driving CF pulmonary disease, we discuss (i) the progression of disease within the primitive CF lung, specifically focusing on the role of host versus bacterial factors; (ii) critical, neutrophil-derived innate immune effectors that are implicated in CF pulmonary disease, including reactive oxygen species (ROS) and antimicrobial peptides (e.g., LL-37); (iii) P. aeruginosa virulence factors and adaptive mutations that enable evasion of the host response; and (iv) ongoing work examining the distribution and colocalization of host and bacterial factors within distinct anatomical niches of the CF lung.
Keywords: ROS; airway; antimicrobial peptides; cystic fibrosis; inflammation; innate immunity; lung infection; reactive oxygen species.
Copyright © 2019 American Society for Microbiology.
Figures

References
-
- Cystic Fibrosis Foundation. 2017. Cystic Fibrosis Foundation patient registry. 2016 annual data report. Cystic Fibrosis Foundation, Bethesda, MD.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
