

Growth dynamics in naturally progressing chronic lymphocytic leukaemia
- PMID: 31142838
- PMCID: PMC6630176
- DOI: 10.1038/s41586-019-1252-x
Growth dynamics in naturally progressing chronic lymphocytic leukaemia
Abstract
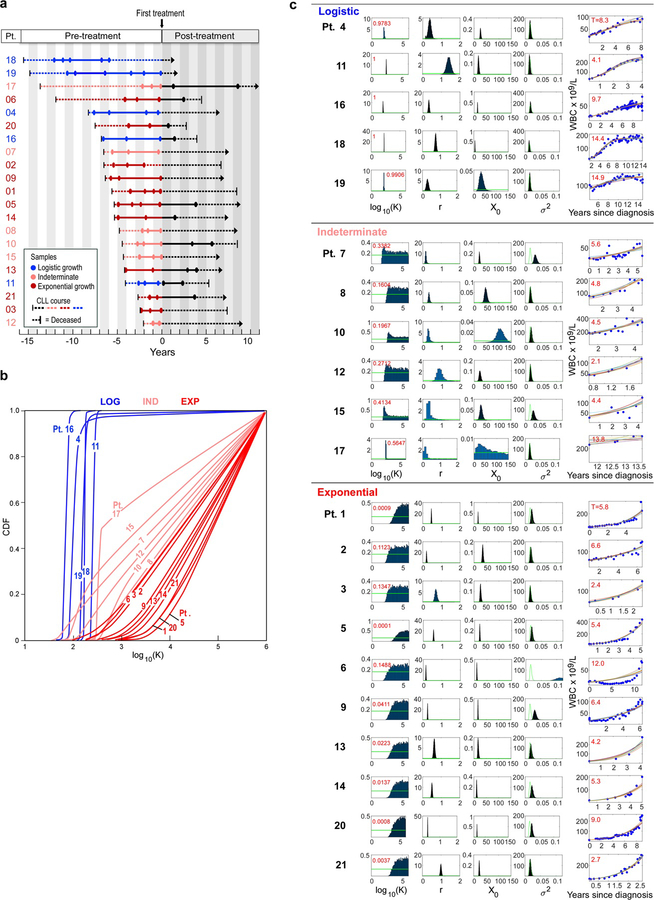
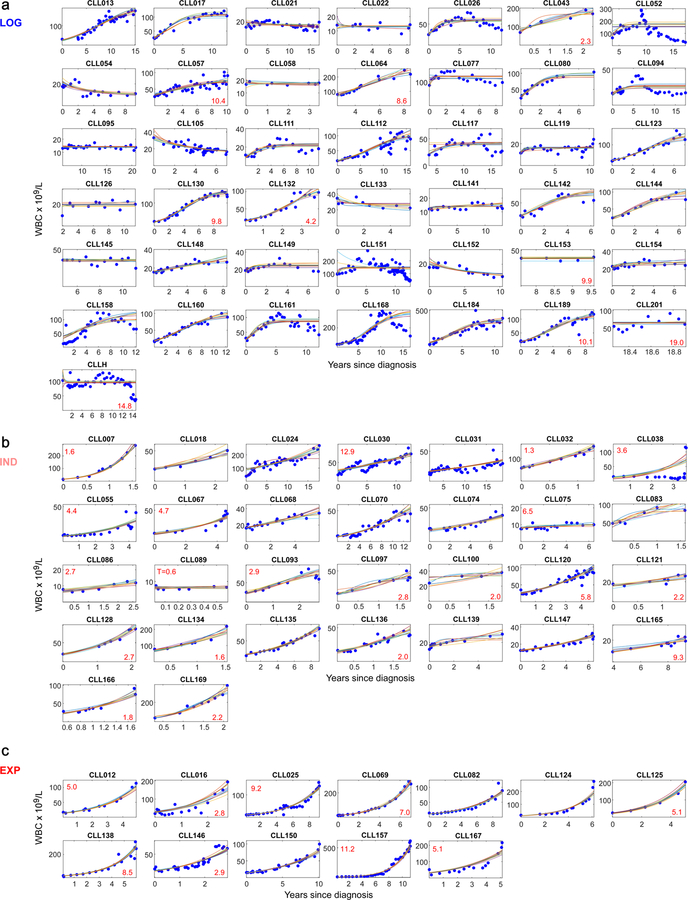
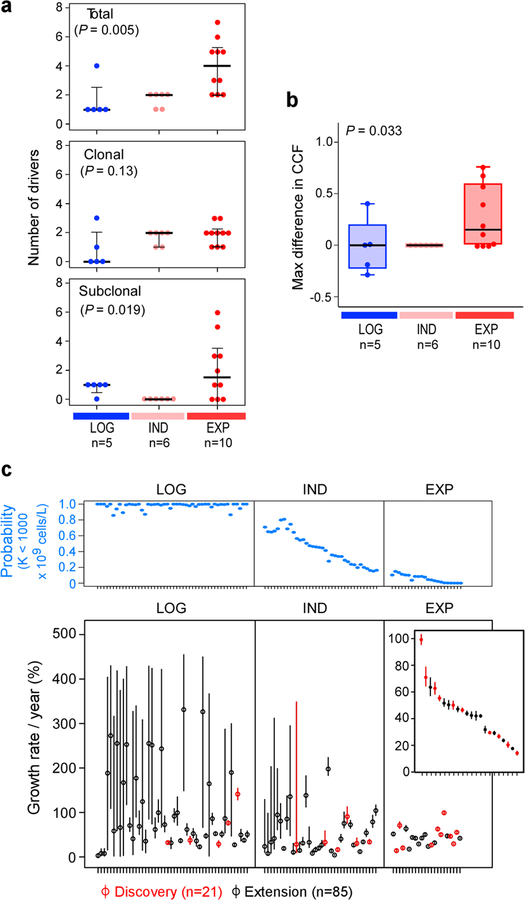
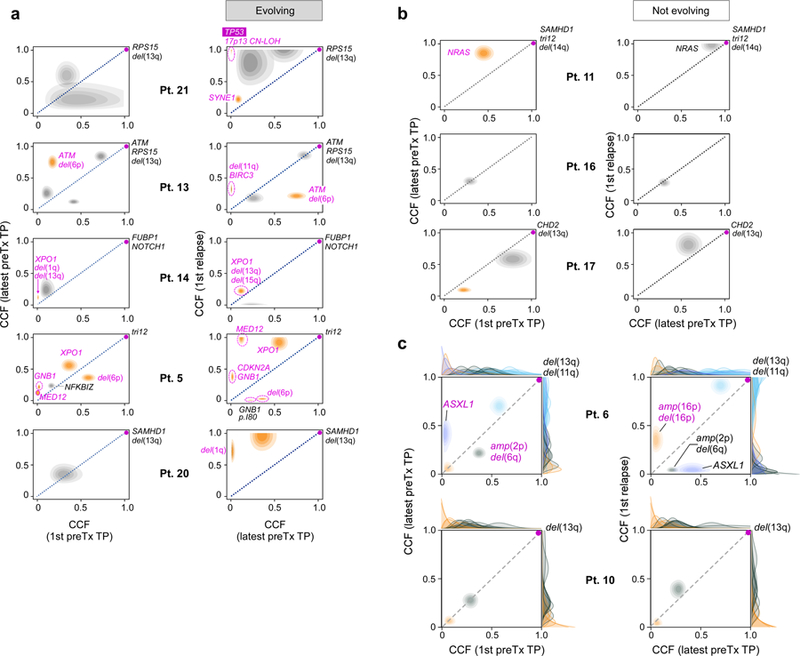
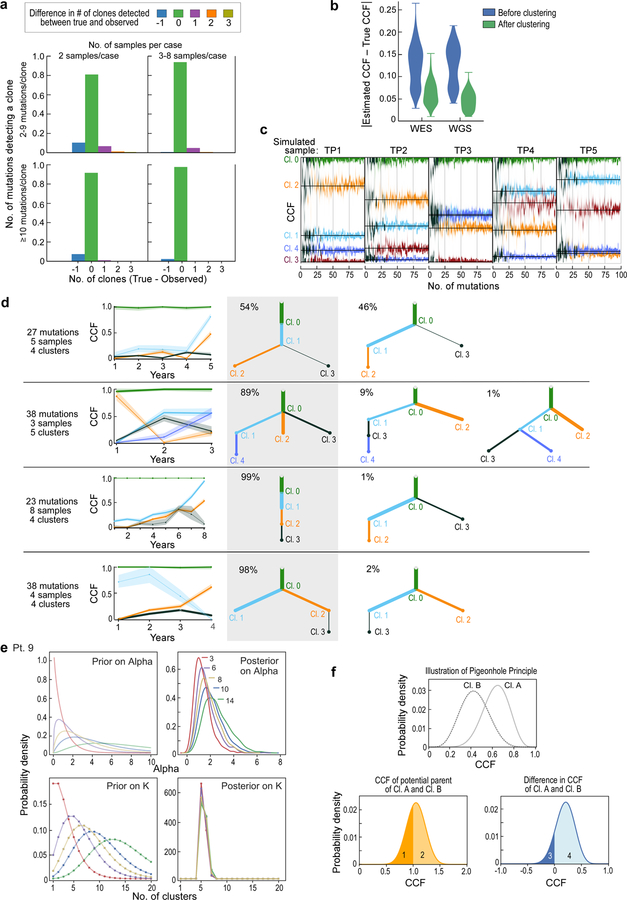
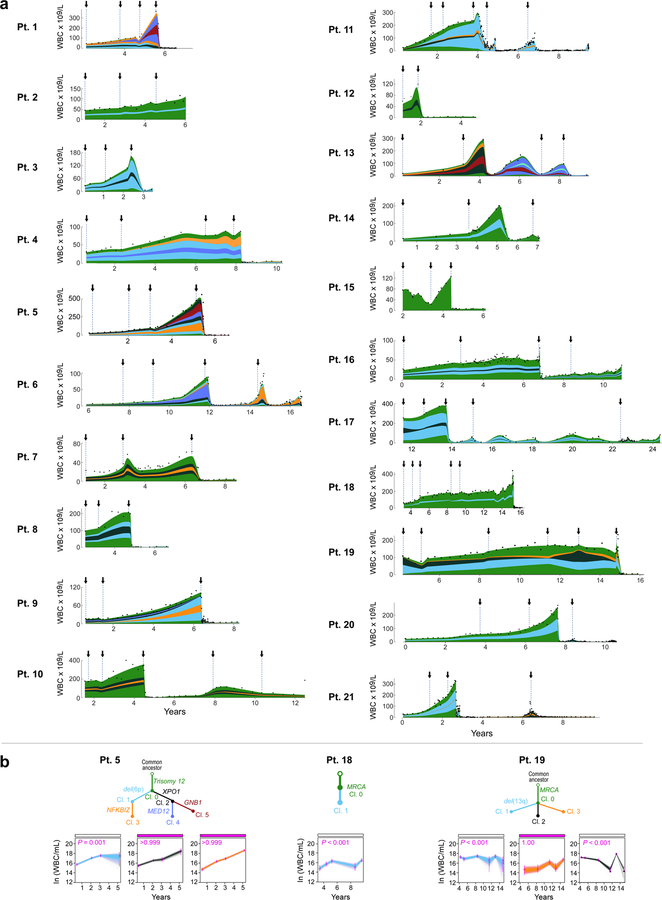
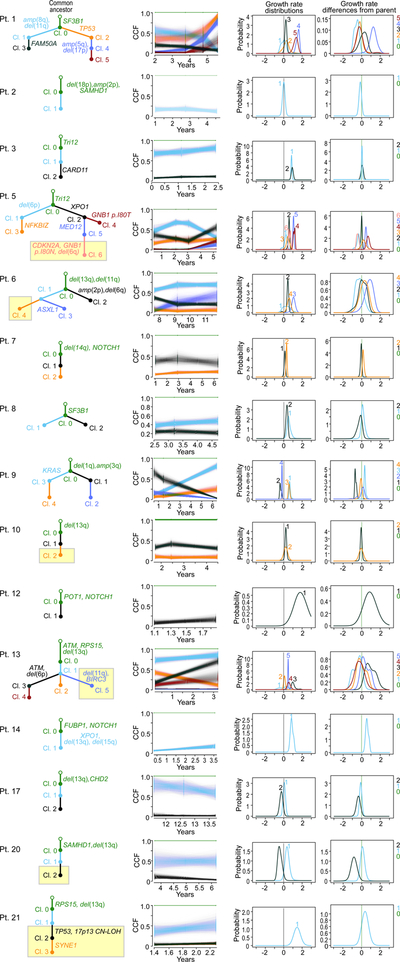
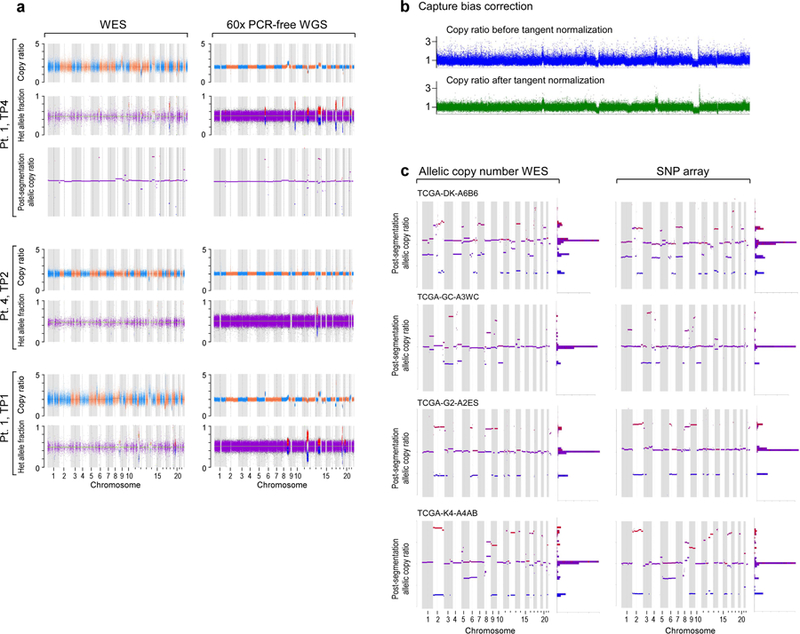
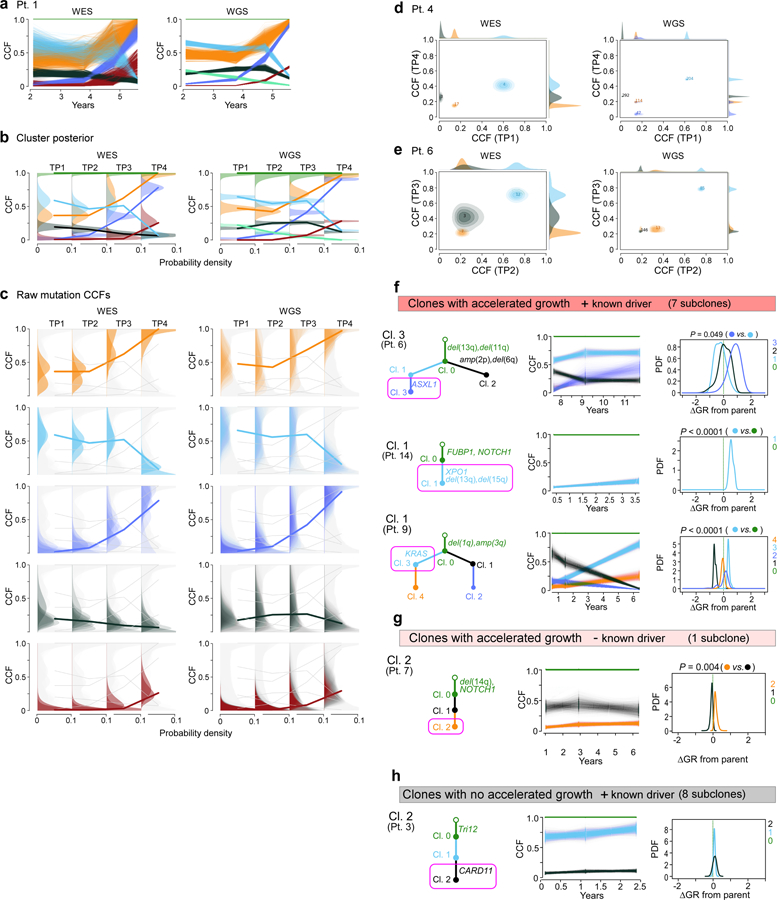
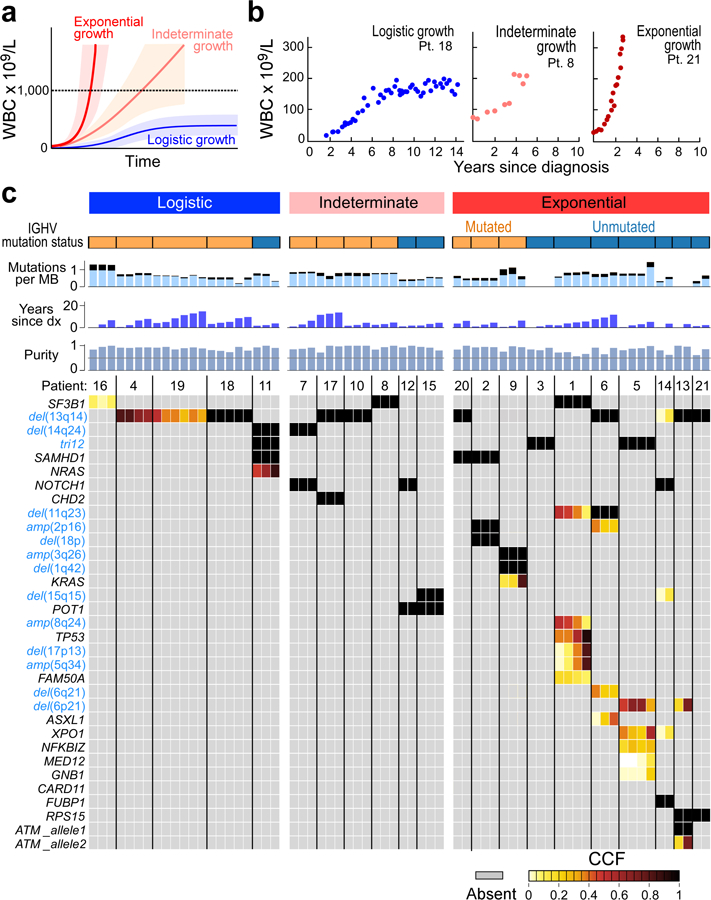
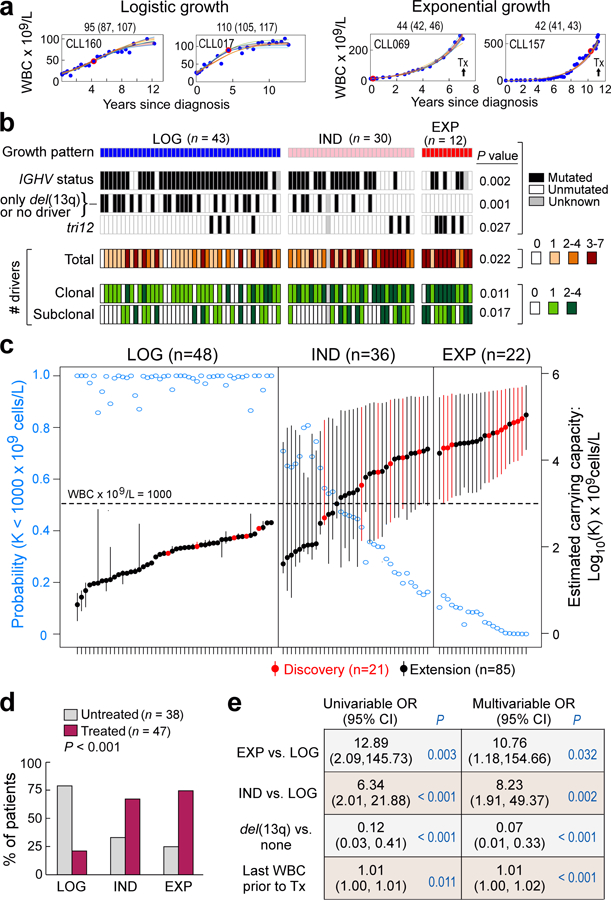
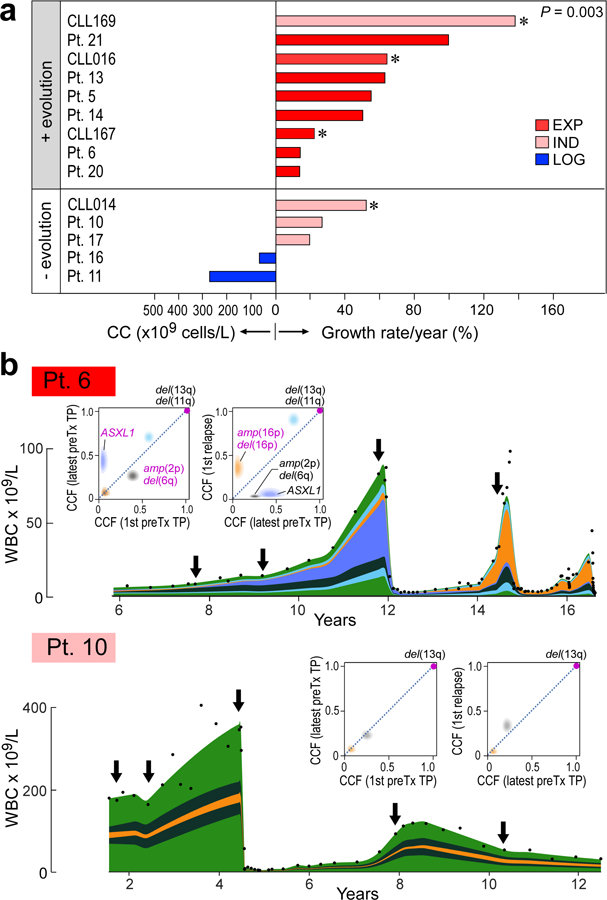
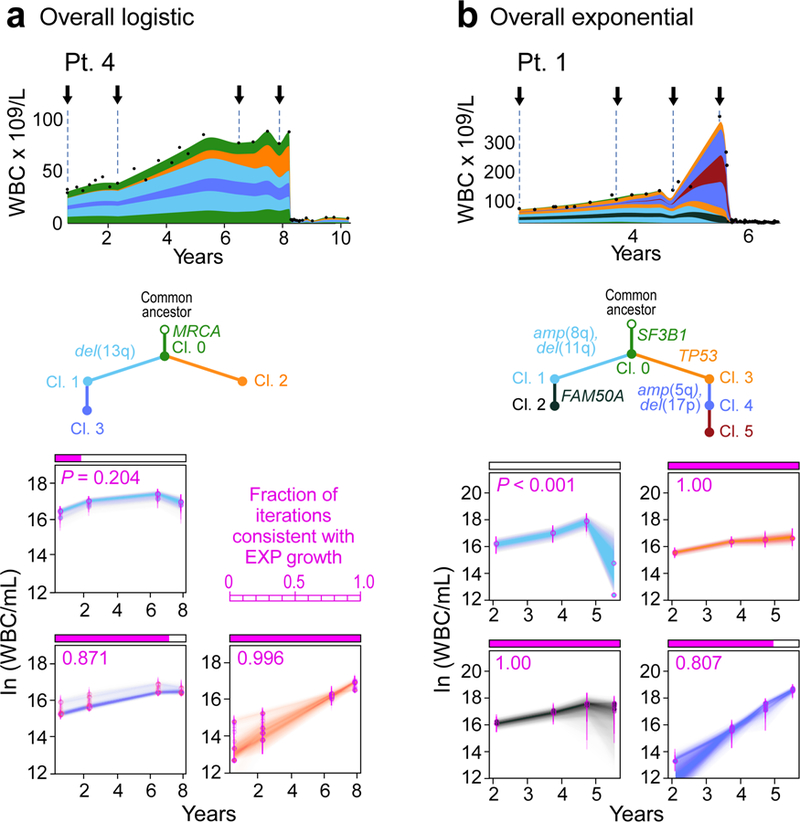
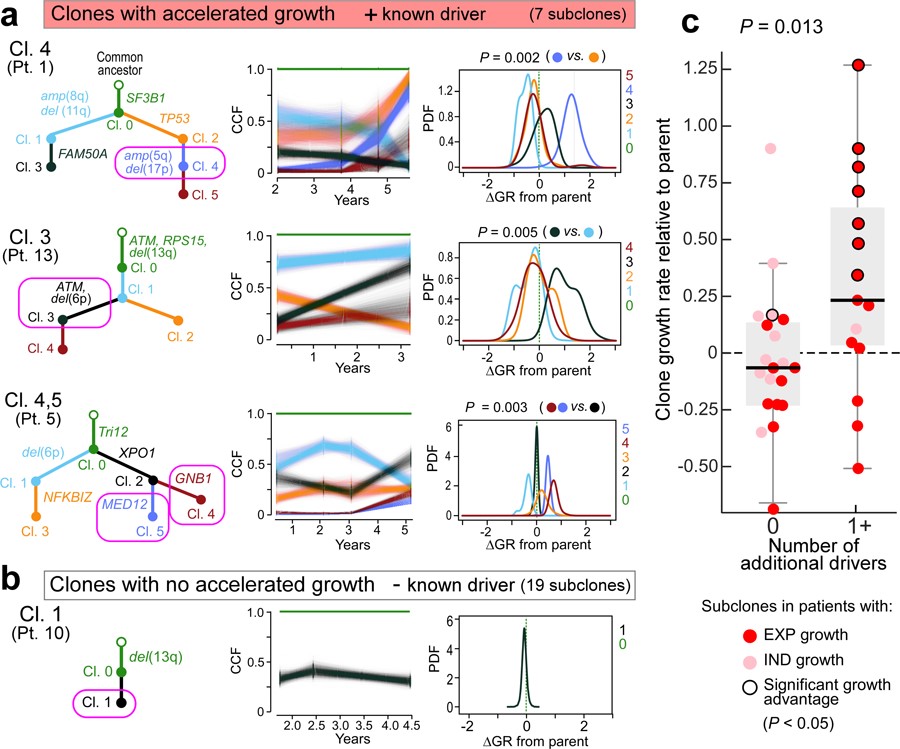
How the genomic features of a patient's cancer relate to individual disease kinetics remains poorly understood. Here we used the indolent growth dynamics of chronic lymphocytic leukaemia (CLL) to analyse the growth rates and corresponding genomic patterns of leukaemia cells from 107 patients with CLL, spanning decades-long disease courses. We found that CLL commonly demonstrates not only exponential expansion but also logistic growth, which is sigmoidal and reaches a certain steady-state level. Each growth pattern was associated with marked differences in genetic composition, the pace of disease progression and the extent of clonal evolution. In a subset of patients, whose serial samples underwent next-generation sequencing, we found that dynamic changes in the disease course of CLL were shaped by the genetic events that were already present in the early slow-growing stages. Finally, by analysing the growth rates of subclones compared with their parental clones, we quantified the growth advantage conferred by putative CLL drivers in vivo.
Conflict of interest statement
Competing interests
C.J.W. is founder of Neon Therapeutics and a member of its scientific advisory board. G.G. receives research funds from IBM and Pharmacyclics. G.G is an inventor of several bioinformatics-related patents, including related to MuTect and ABSOLUTE. C.J.W., D.N. and T.J.K. receive research funding from Pharmacyclics. J.S. is a current employee of Moderna Therapeutics. J.G.G. receives grant funding from funding Janssen, Acerta, Celgene; Honoraria Abbvie, AZ, Celgene, Kite, Janssen, Pharmacyclics, Roche and Novartis. K.R.R. is on Medical Advisory Boards of Pharmacyclics, Roche/Genentech, and Cellectis. J.R.B. is a consultant for Abbvie, Acerta, Beigene, Genentech/Roche, Gilead, Juno/Celgene, Kite, Loxo, Novartis, Pfizer, Pharmacyclics, Sunesis, TG Therapeutics, Verastem; received honoraria from Janssen and Teva; received research funding from Gilead, Loxo, Sun and Verastem; and served on data safety monitoring committees for Morphosys and Invectys. The other authors declare no potential conflicts of interest.
Figures
References
-
- Wodarz D & Komarova NL Dynamics of cancer : mathematical foundations of oncology (World Scientific, 2014).
-
- Norton L A Gompertzian model of human breast cancer growth. Cancer Res 48, 7067–7071 (1988). - PubMed
-
- Spratt JA, von Fournier D, Spratt JS & Weber EE Decelerating growth and human breast cancer. Cancer 71, 2013–2019 (1993). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
