

Flexible and scalable diagnostic filtering of genomic variants using G2P with Ensembl VEP
- PMID: 31147538
- PMCID: PMC6542828
- DOI: 10.1038/s41467-019-10016-3
Flexible and scalable diagnostic filtering of genomic variants using G2P with Ensembl VEP
Abstract
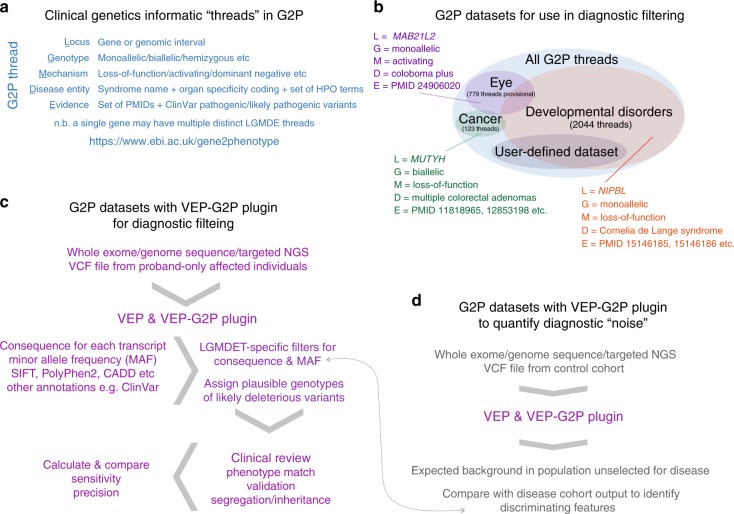
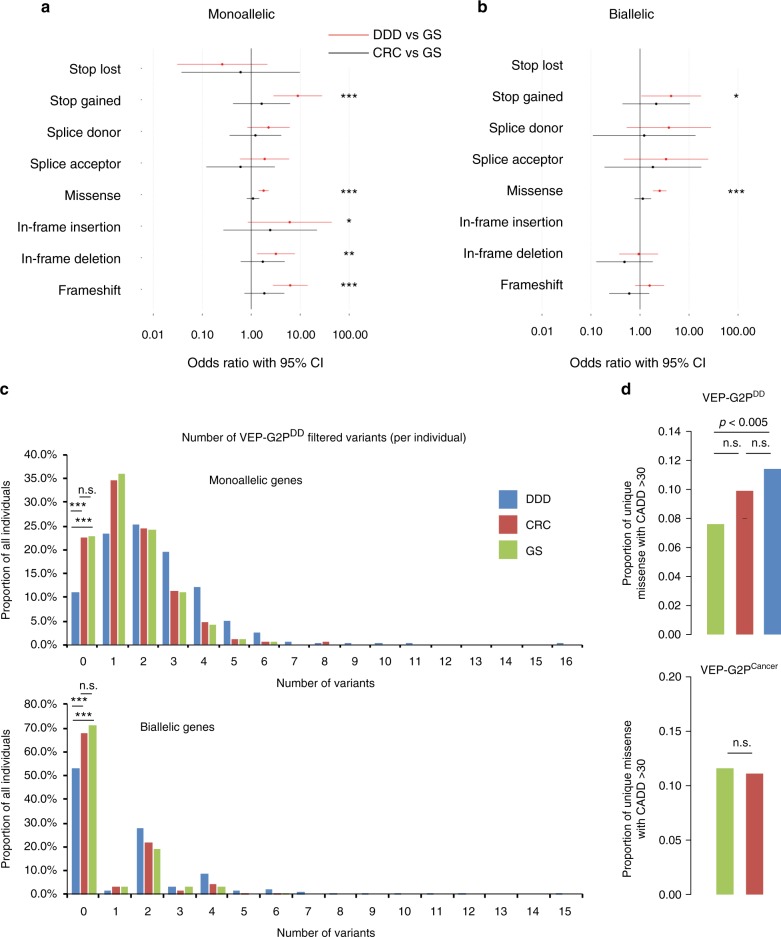
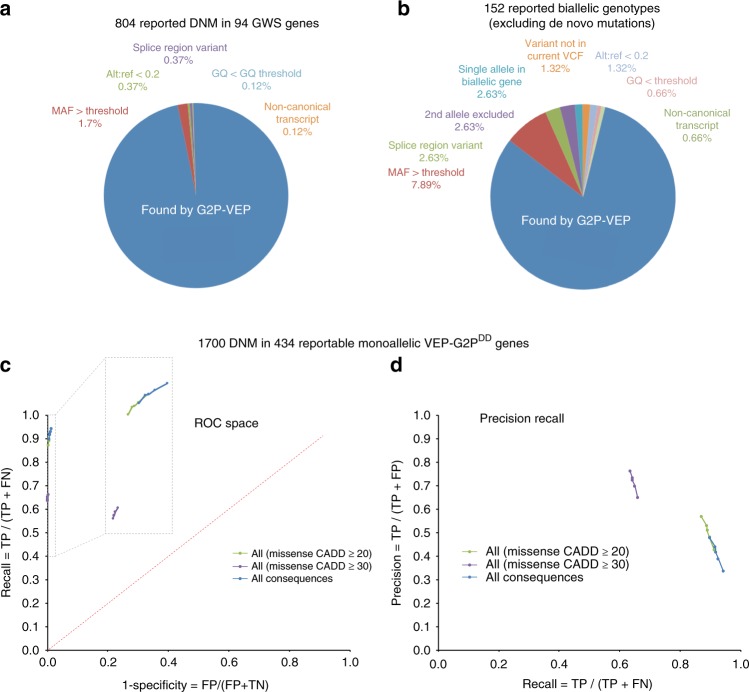
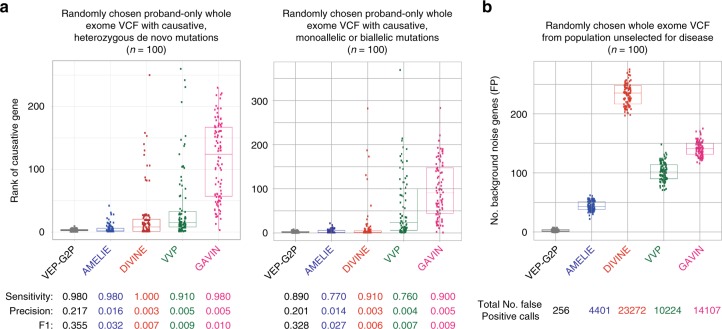
We aimed to develop an efficient, flexible and scalable approach to diagnostic genome-wide sequence analysis of genetically heterogeneous clinical presentations. Here we present G2P ( www.ebi.ac.uk/gene2phenotype ) as an online system to establish, curate and distribute datasets for diagnostic variant filtering via association of allelic requirement and mutational consequence at a defined locus with phenotypic terms, confidence level and evidence links. An extension to Ensembl Variant Effect Predictor (VEP), VEP-G2P was used to filter both disease-associated and control whole exome sequence (WES) with Developmental Disorders G2P (G2PDD; 2044 entries). VEP-G2PDD shows a sensitivity/precision of 97.3%/33% for de novo and 81.6%/22.7% for inherited pathogenic genotypes respectively. Many of the missing genotypes are likely false-positive pathogenic assignments. The expected number and discriminative features of background genotypes are defined using control WES. Using only human genetic data VEP-G2P performs well compared to other freely-available diagnostic systems and future phenotypic matching capabilities should further enhance performance.
Conflict of interest statement
M.E.H. is a co-founder, consultant and non-executive director of Congenica Ltd. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
