

Understanding histone H3 lysine 36 methylation and its deregulation in disease
- PMID: 31147750
- PMCID: PMC11105573
- DOI: 10.1007/s00018-019-03144-y
Understanding histone H3 lysine 36 methylation and its deregulation in disease
Abstract
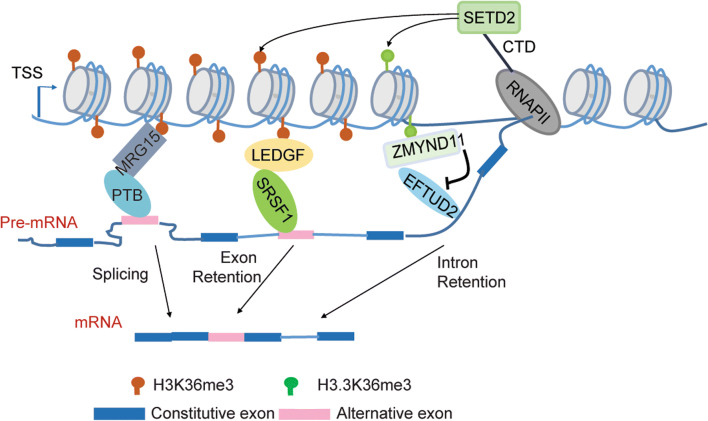
Methylation of histone H3 lysine 36 (H3K36) plays crucial roles in the partitioning of chromatin to distinctive domains and the regulation of a wide range of biological processes. Trimethylation of H3K36 (H3K36me3) demarcates body regions of the actively transcribed genes, providing signals for modulating transcription fidelity, mRNA splicing and DNA damage repair; and di-methylation of H3K36 (H3K36me2) spreads out within large intragenic regions, regulating distribution of histone H3 lysine 27 trimethylation (H3K27me3) and possibly DNA methylation. These H3K36 methylation-mediated events are biologically crucial and controlled by different classes of proteins responsible for either 'writing', 'reading' or 'erasing' of H3K36 methylation marks. Deregulation of H3K36 methylation and related regulatory factors leads to pathogenesis of disease such as developmental syndrome and cancer. Additionally, recurrent mutations of H3K36 and surrounding histone residues are detected in human tumors, further highlighting the importance of H3K36 in biology and medicine. This review will elaborate on current advances in understanding H3K36 methylation and related molecular players during various chromatin-templated cellular processes, their crosstalks with other chromatin factors, as well as their deregulations in the diseased contexts.
Keywords: Cancer; Chromatin; DNA damage repair; DNA methylation; Demethylase; Epigenetics; Gene transcription; H3K27me3; H3K36 methylation; H3K36me2; H3K36me3; Histone modification; Methyltransferase; Splicing.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
