

Genotyping genetically heterogeneous Cyclospora cayetanensis infections to complement epidemiological case linkage
- PMID: 31148531
- PMCID: PMC6699905
- DOI: 10.1017/S0031182019000581
Genotyping genetically heterogeneous Cyclospora cayetanensis infections to complement epidemiological case linkage
Abstract
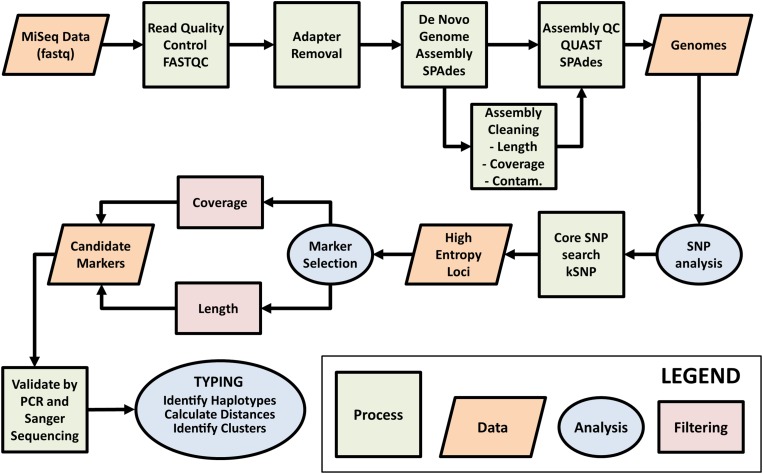
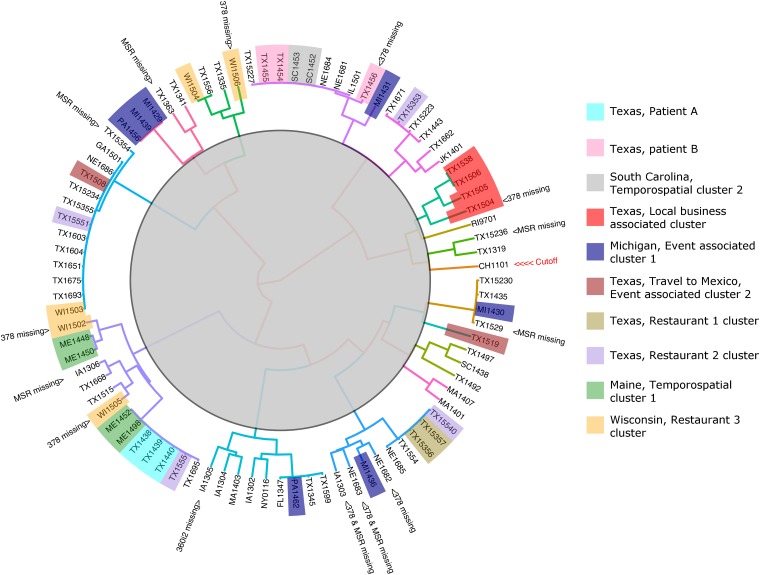
Sexually reproducing pathogens such as Cyclospora cayetanensis often produce genetically heterogeneous infections where the number of unique sequence types detected at any given locus varies depending on which locus is sequenced. The genotypes assigned to these infections quickly become complex when additional loci are analysed. This genetic heterogeneity confounds the utility of traditional sequence-typing and phylogenetic approaches for aiding epidemiological trace-back, and requires new methods to address this complexity. Here, we describe an ensemble of two similarity-based classification algorithms, including a Bayesian and heuristic component that infer the relatedness of C. cayetanensis infections. The ensemble requires a set of haplotypes as input and assigns arbitrary distances to specimen pairs reflecting their most likely relationships. The approach was applied to data generated from a test cohort of 88 human fecal specimens containing C. cayetanensis, including 30 from patients whose infections were associated with epidemiologically defined outbreak clusters of cyclosporiasis. The ensemble assigned specimens to plausible clusters of genetically related infections despite their complex haplotype composition. These relationships were corroborated by a significant number of epidemiological linkages (P < 0.0001) suggesting the ensemble's utility for aiding epidemiological trace-back investigations of cyclosporiasis.
Keywords: Algorithm; Bayesian; Cyclospora cayetanensis; bioinformatics; ensemble; epidemiology; heuristic.
Figures

References
-
- Abanyie F, Harvey RR, Harris JR, Wiegand RE, Gaul L, Desvignes-Kendrick M, Irvin K, Williams I, Hall RL, Herwaldt B, Gray EB, Qvarnstrom Y, Wise ME, Cantu V, Cantey PT, Bosch S, AJ DAS, Fields A, Bishop H, Wellman A, Beal J, Wilson N, Fiore AE, Tauxe R, Lance S, Slutsker L, Parise M and Multistate Cyclosporiasis Outbreak Investigation Team (2015) 2013 Multistate outbreaks of Cyclospora cayetanensis infections associated with fresh produce: focus on the Texas investigations. Epidemiology and Infection 143, 3451–3458. - PMC - PubMed
-
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA and Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19, 455–477. - PMC - PubMed
-
- CDC (2017a) Cyclosporiasis: Outbreak Investigations and Updates. Vol. 2017 Centers for Disease Control and Prevention, Global Health, Division of Parasitic Disease, Atlanta, Georgia, USA https://www.cdc.gov/parasites/cyclosporiasis/outbreaks/index.html
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
