

Molecular Vertical Excitation Energies Studied with First-Order RASSCF (RAS[1,1]): Balancing Covalent and Ionic Excited States
- PMID: 31150228
- PMCID: PMC7007262
- DOI: 10.1021/acs.jpca.9b03715
Molecular Vertical Excitation Energies Studied with First-Order RASSCF (RAS[1,1]): Balancing Covalent and Ionic Excited States
Abstract
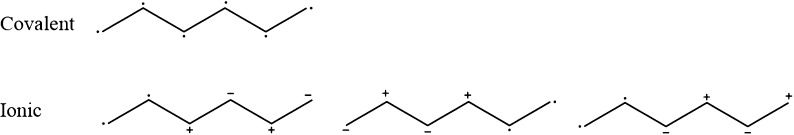
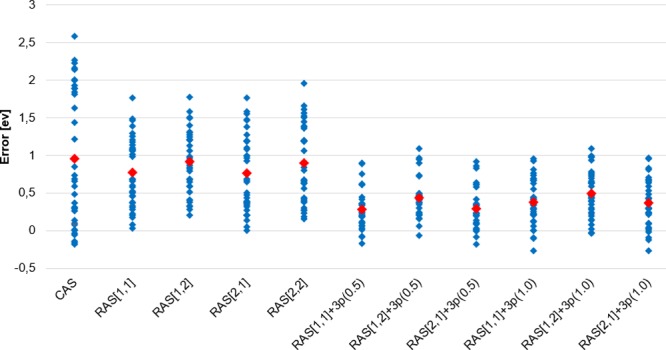
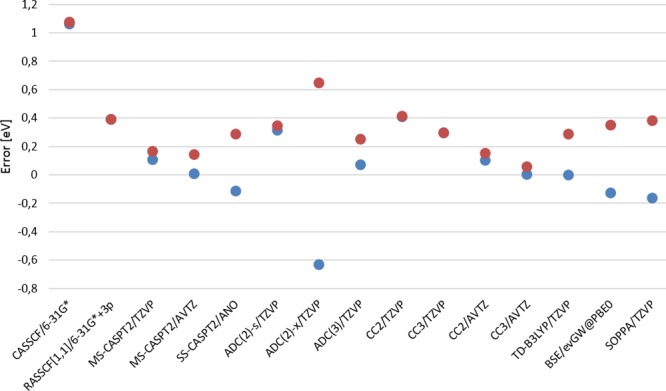
RASSCF calculations of vertical excitation energies were carried out on a benchmark set of 19 organic molecules studied by Thiel and co-workers [ J. Chem. Phys. 2008 , 128 , 134110 ]. The best results, in comparison with the MS-CASPT2 results of Thiel, were obtained using a RASSCF space that contains at most one hole and one particle in the RAS1 and RAS3 spaces, respectively, which we denote as RAS[1,1]. This subset of configurations recovers mainly the effect of polarization and semi-internal electronic correlation that is only included in CASSCF in an averaged way. Adding all-external correlation by allowing double excitations from RAS1 and RAS2 into RAS3 did not improve the results, and indeed, they were slightly worse. The accuracy of the first-order RASSCF computations is demonstrated to be a function of whether the state of interest can be classified as covalent or ionic in the space of configurations built from orbitals localized onto atomic sites. For covalent states, polarization and semi-internal correlation effects are negligible (RAS[1,1]), while for ionic states, these effects are large (because of inherent diffusiveness of these states compared to the covalent states) and, thus, an acceptable agreement with MS-CASPT2 can be obtained using first-order RASSCF with the extra basis set involving 3p orbitals in most cases. However, for those ionic states that are quasi-degenerate with a Rydberg state or for nonlocal nπ* states, there remains a significant error resulting from all external correlation effects.
Conflict of interest statement
The authors declare no competing financial interest.
Figures




References
-
- Robb M. A.Theoretical Chemistry for Electronic Excited States; Theoretical and Computational Chemistry Series; The Royal Society of Chemistry, 2018. (10.1039/9781788013642) - DOI
-
- Löwdin P. O. Quantum Theory of Many-Particle Systems. III. Extension of the Hartree-Fock Scheme to Include Degenerate Systems and Correlation Effects. Phys. Rev. 1955, 97, 1509–1520. 10.1103/PhysRev.97.1509. - DOI
-
- Clotet A.; Daudey J. P.; Malrieu J. P.; Rubio J.; Spiegelmann F. The Effect of Dynamical Correlation on the Valence Wavefunction of Molecules: Dressed Complete Active Space Self-Consistent Field Calculations. Chem. Phys. 1990, 147, 293–307. 10.1016/0301-0104(90)85045-X. - DOI
-
- Karafiloglou P.; Malrieu J. P. The Effect of Electronic Correlation on Molecular Wavefunctions. Chem. Phys. 1986, 104, 383–398. 10.1016/0301-0104(86)85027-3. - DOI
-
- Roos B. O.; Taylor P. R. A Complete Active Space SCF Method (CASSCF) Using A Density Matrix Formulated Super-CI Approach. Chem. Phys. 1980, 48, 157–173. 10.1016/0301-0104(80)80045-0. - DOI
