

EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome
- PMID: 31151987
- PMCID: PMC6754720
- DOI: 10.1182/blood.2018893404
EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome
Abstract
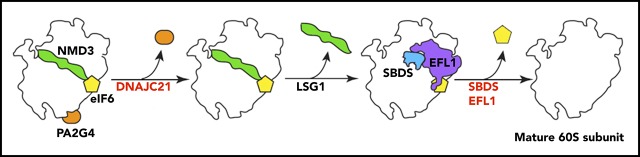
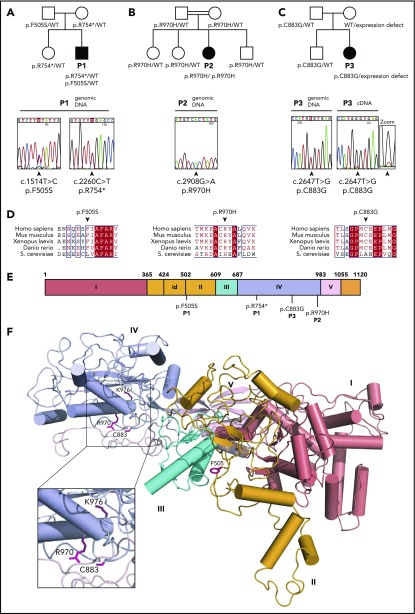
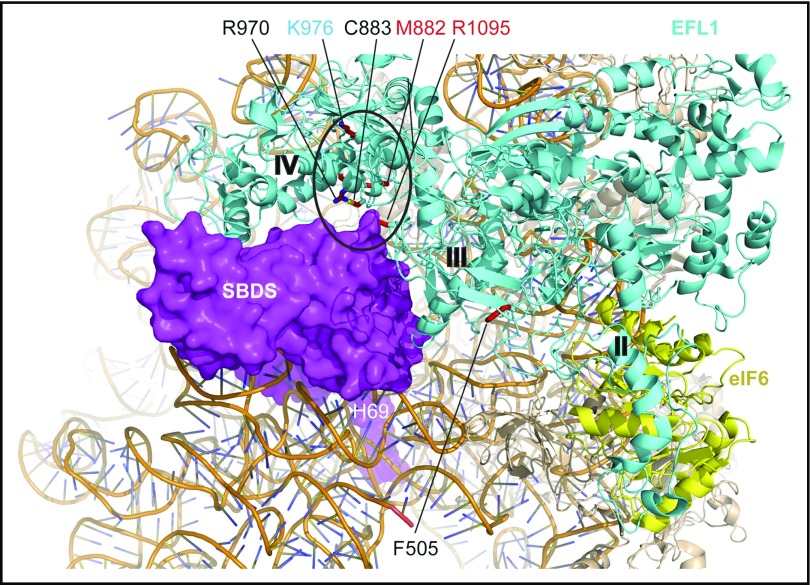
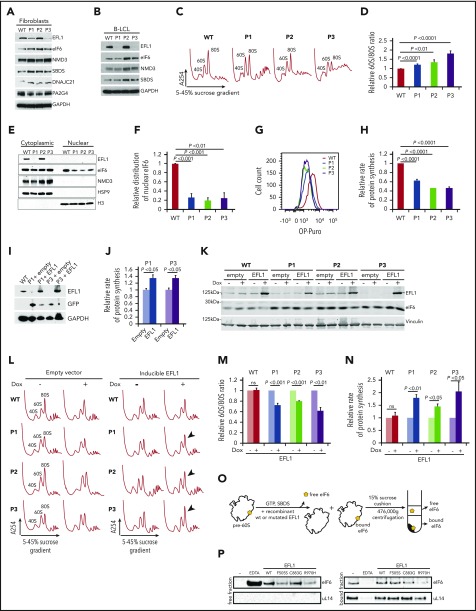
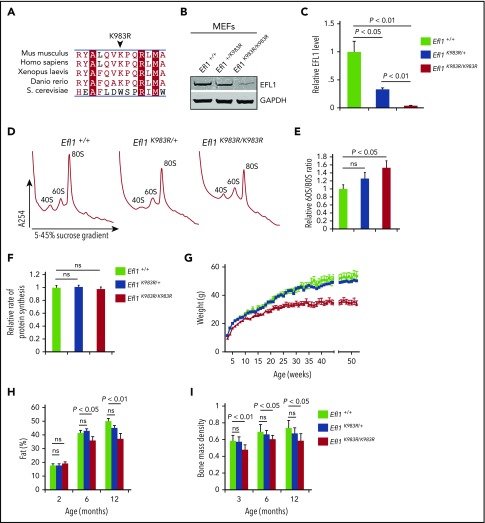
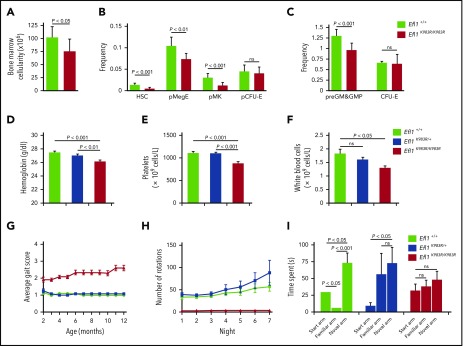
Shwachman-Diamond syndrome (SDS) is a recessive disorder typified by bone marrow failure and predisposition to hematological malignancies. SDS is predominantly caused by deficiency of the allosteric regulator Shwachman-Bodian-Diamond syndrome that cooperates with elongation factor-like GTPase 1 (EFL1) to catalyze release of the ribosome antiassociation factor eIF6 and activate translation. Here, we report biallelic mutations in EFL1 in 3 unrelated individuals with clinical features of SDS. Cellular defects in these individuals include impaired ribosomal subunit joining and attenuated global protein translation as a consequence of defective eIF6 eviction. In mice, Efl1 deficiency recapitulates key aspects of the SDS phenotype. By identifying biallelic EFL1 mutations in SDS, we define this leukemia predisposition disorder as a ribosomopathy that is caused by corruption of a fundamental, conserved mechanism, which licenses entry of the large ribosomal subunit into translation.
© 2019 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
References
-
- Donadieu J, Fenneteau O, Beaupain B, et al. ; Associated investigators of the French Severe Chronic Neutropenia Registry* . Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica. 2012;97(9):1312-1319. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
