

Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer
- PMID: 31152137
- PMCID: PMC6589752
- DOI: 10.1073/pnas.1818521116
Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer
Abstract
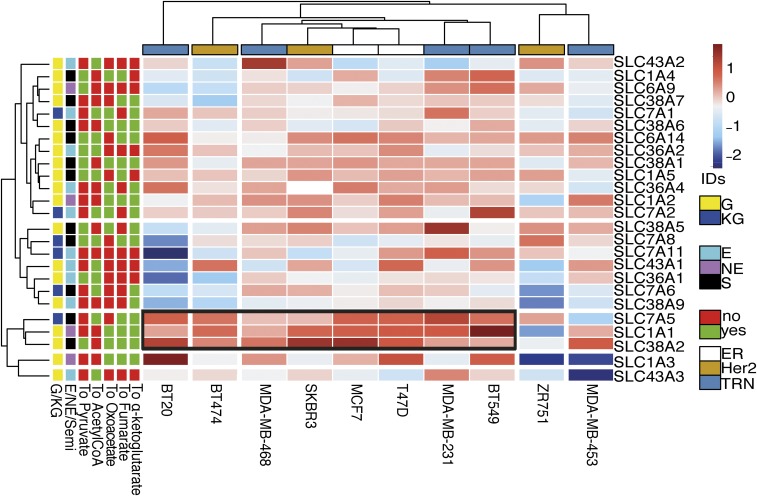
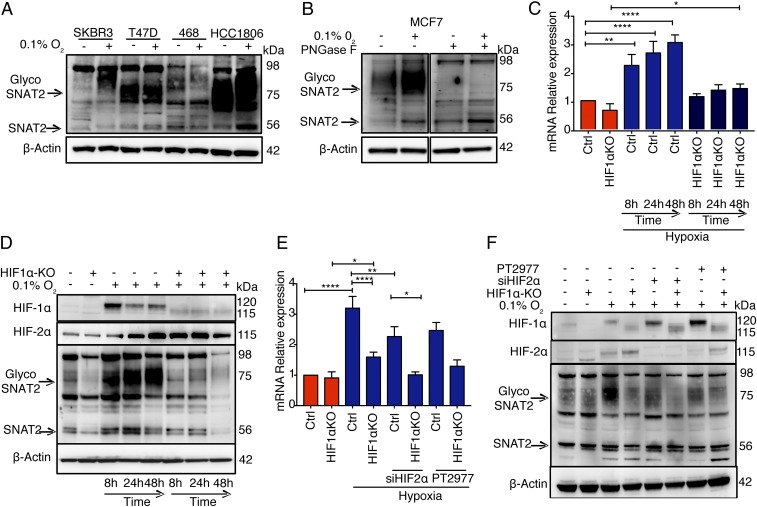
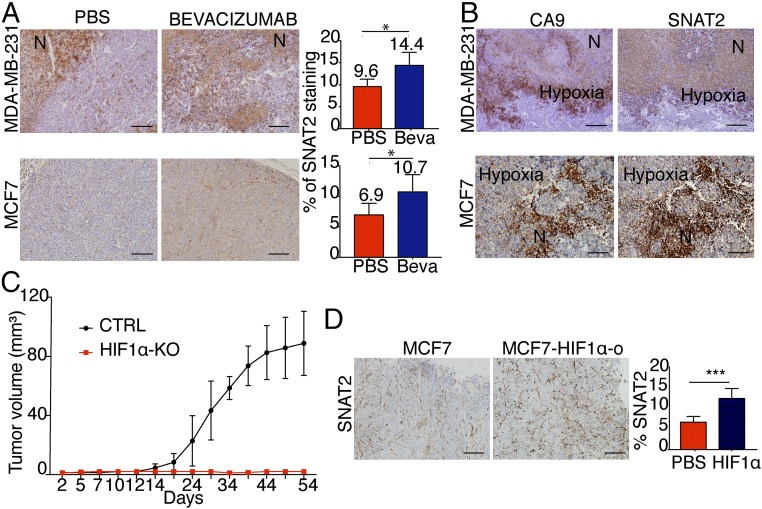
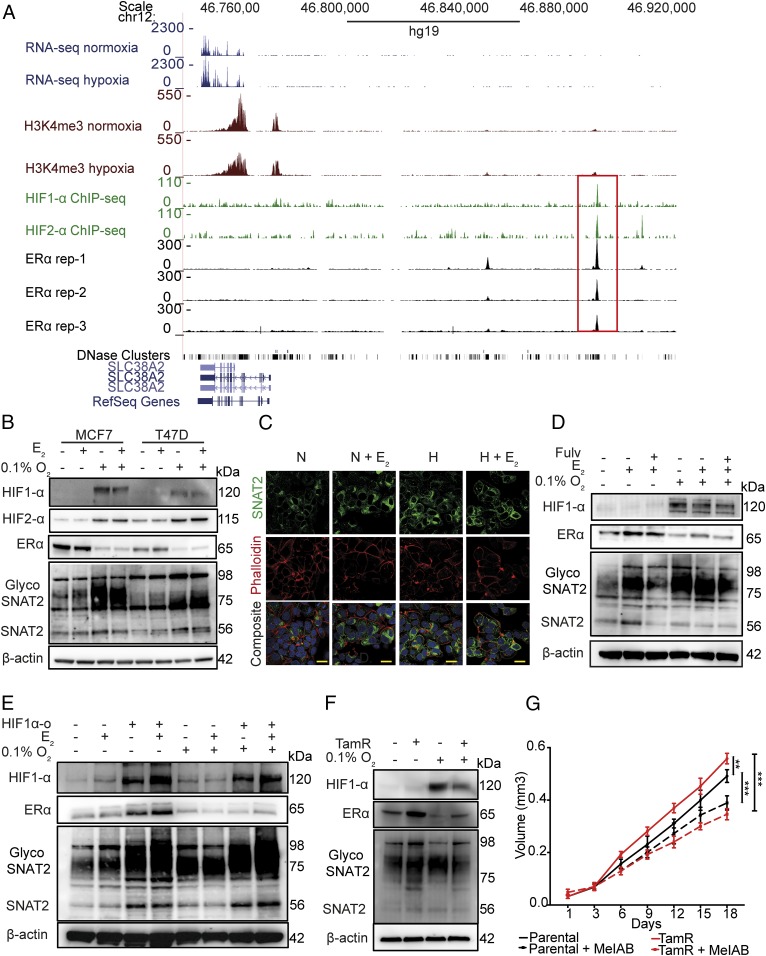
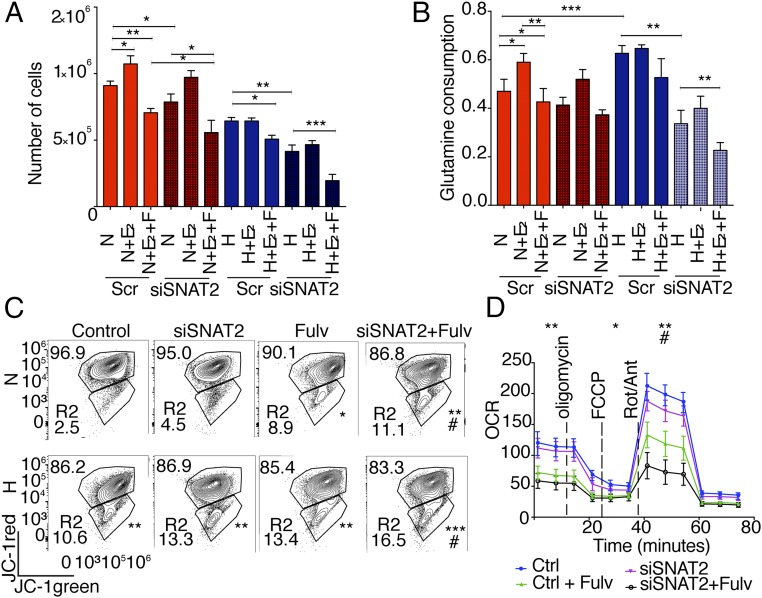
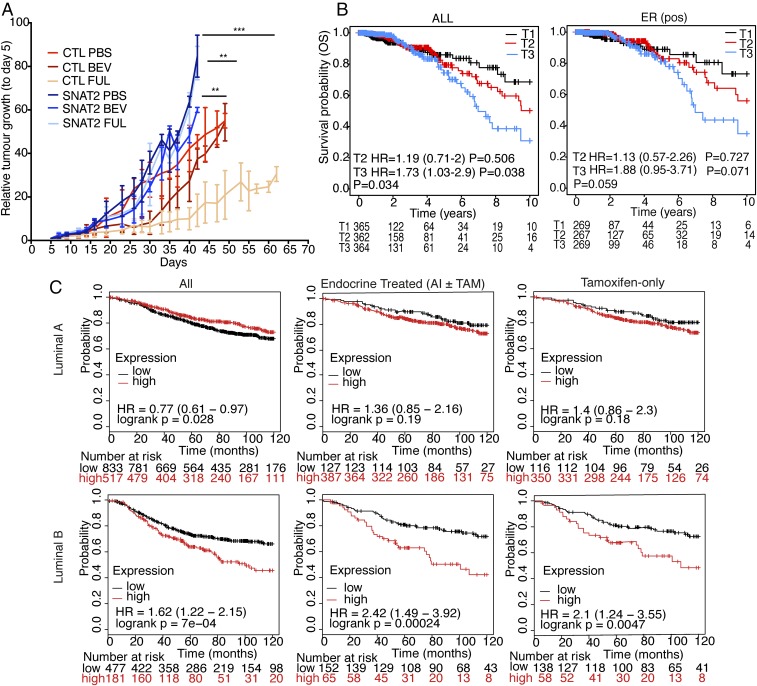
Tumor hypoxia is associated with poor patient outcomes in estrogen receptor-α-positive (ERα+) breast cancer. Hypoxia is known to affect tumor growth by reprogramming metabolism and regulating amino acid (AA) uptake. Here, we show that the glutamine transporter, SNAT2, is the AA transporter most frequently induced by hypoxia in breast cancer, and is regulated by hypoxia both in vitro and in vivo in xenografts. SNAT2 induction in MCF7 cells was also regulated by ERα, but it became predominantly a hypoxia-inducible factor 1α (HIF-1α)-dependent gene under hypoxia. Relevant to this, binding sites for both HIF-1α and ERα overlap in SNAT2's cis-regulatory elements. In addition, the down-regulation of SNAT2 by the ER antagonist fulvestrant was reverted in hypoxia. Overexpression of SNAT2 in vitro to recapitulate the levels induced by hypoxia caused enhanced growth, particularly after ERα inhibition, in hypoxia, or when glutamine levels were low. SNAT2 up-regulation in vivo caused complete resistance to antiestrogen and, partially, anti-VEGF therapies. Finally, high SNAT2 expression levels correlated with hypoxia profiles and worse outcome in patients given antiestrogen therapies. Our findings show a switch in the regulation of SNAT2 between ERα and HIF-1α, leading to endocrine resistance in hypoxia. Development of drugs targeting SNAT2 may be of value for a subset of hormone-resistant breast cancer.
Keywords: ERα; amino acid transporter; breast cancer; cancer metabolism; hypoxia.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2017. CA Cancer J. Clin. 67, 7–30 (2017). - PubMed
-
- Harbeck N., Gnant M., Breast cancer. Lancet 389, 1134–1150 (2017). - PubMed
-
- Tryfonidis K., Zardavas D., Katzenellenbogen B. S., Piccart M., Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat Rev. 50, 68–81 (2016). - PubMed
-
- Musgrove E. A., Sutherland R. L., Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 9, 631–643 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
