

Novel Role for the AnxA1-Fpr2/ALX Signaling Axis as a Key Regulator of Platelet Function to Promote Resolution of Inflammation
- PMID: 31154815
- PMCID: PMC6687438
- DOI: 10.1161/CIRCULATIONAHA.118.039345
Novel Role for the AnxA1-Fpr2/ALX Signaling Axis as a Key Regulator of Platelet Function to Promote Resolution of Inflammation
Erratum in
-
Correction to: Novel Role for the AnxA1-Fpr2/ALX Signaling Axis as a Key Regulator of Platelet Function to Promote Resolution of Inflammation.Circulation. 2019 Jul 23;140(4):e172. doi: 10.1161/CIR.0000000000000713. Epub 2019 Jul 22. Circulation. 2019. PMID: 31329483 Free PMC article. No abstract available.
Abstract
Background: Ischemia reperfusion injury (I/RI) is a common complication of cardiovascular diseases. Resolution of detrimental I/RI-generated prothrombotic and proinflammatory responses is essential to restore homeostasis. Platelets play a crucial part in the integration of thrombosis and inflammation. Their role as participants in the resolution of thromboinflammation is underappreciated; therefore we used pharmacological and genetic approaches, coupled with murine and clinical samples, to uncover key concepts underlying this role.
Methods: Middle cerebral artery occlusion with reperfusion was performed in wild-type or annexin A1 (AnxA1) knockout (AnxA1-/-) mice. Fluorescence intravital microscopy was used to visualize cellular trafficking and to monitor light/dye-induced thrombosis. The mice were treated with vehicle, AnxA1 (3.3 mg/kg), WRW4 (1.8 mg/kg), or all 3, and the effect of AnxA1 was determined in vivo and in vitro.
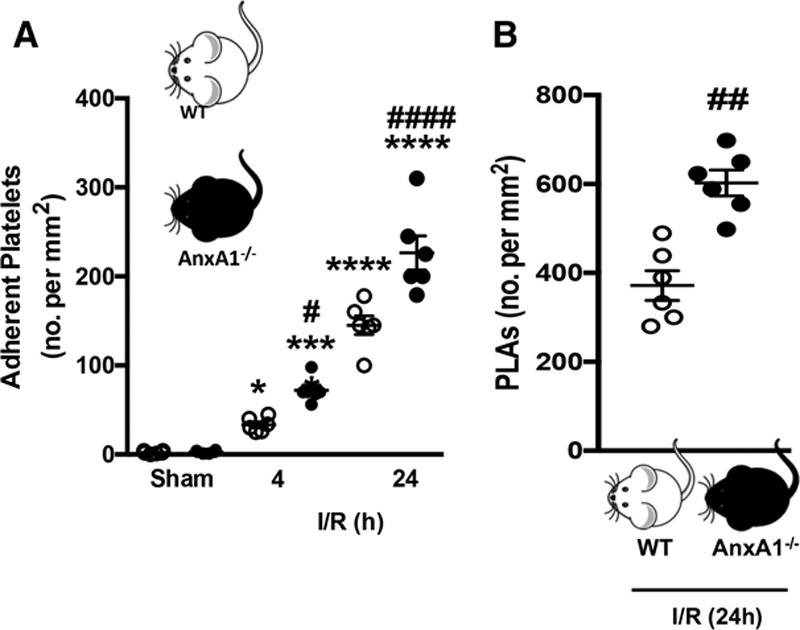
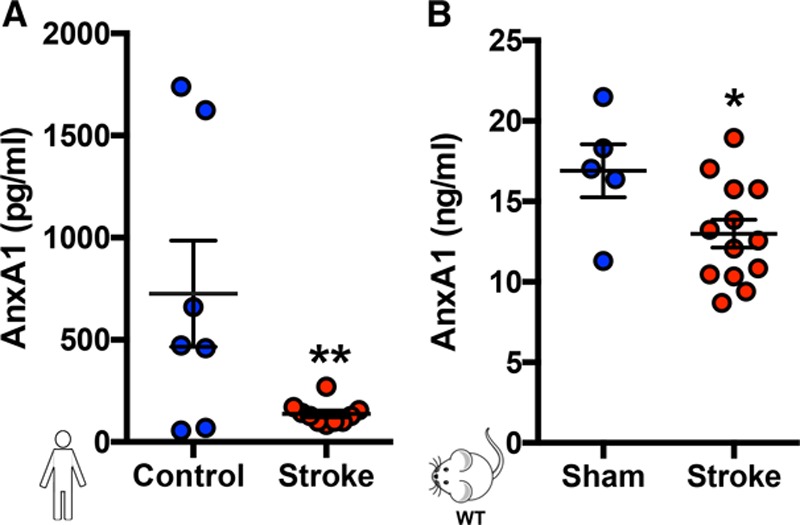
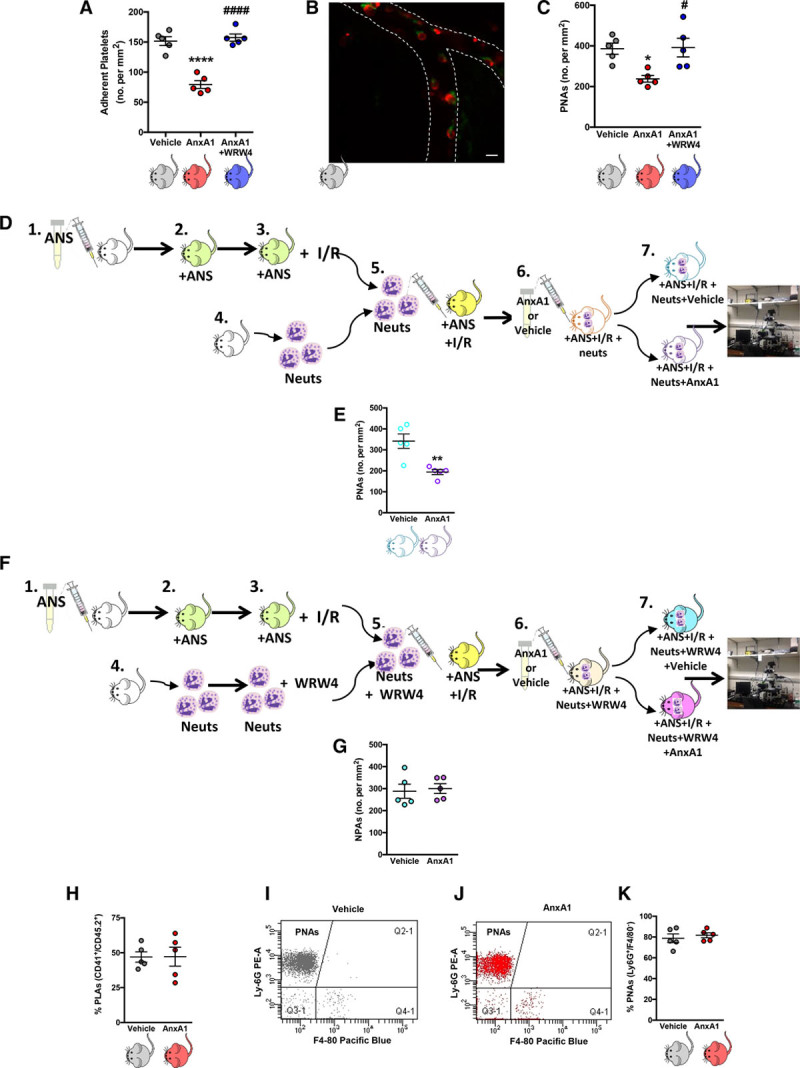
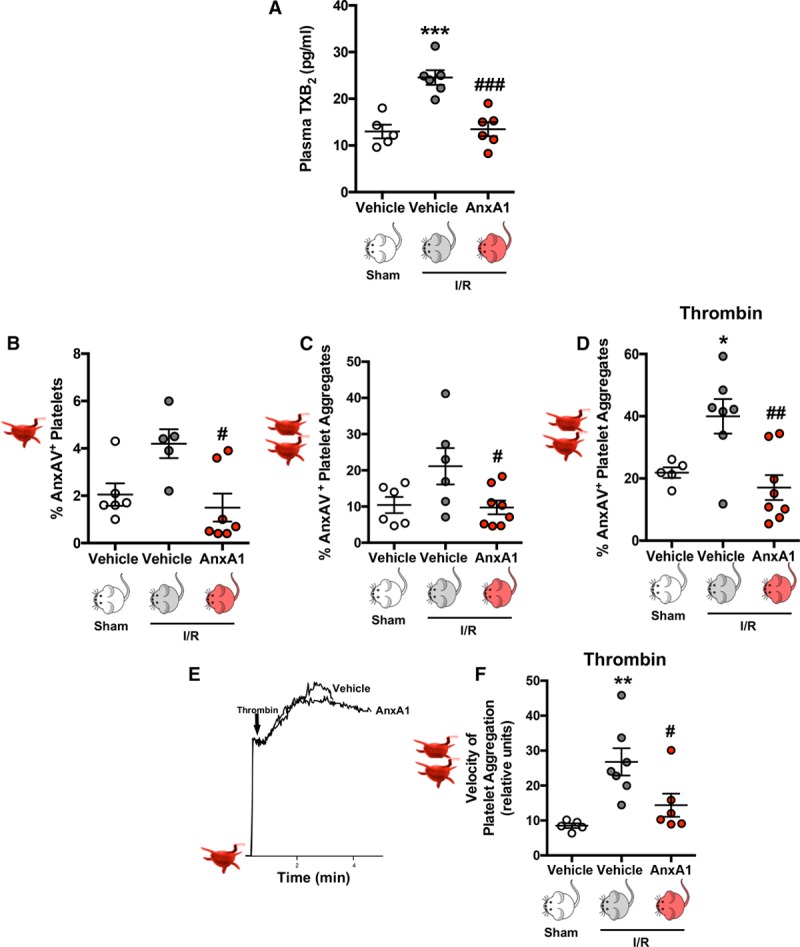
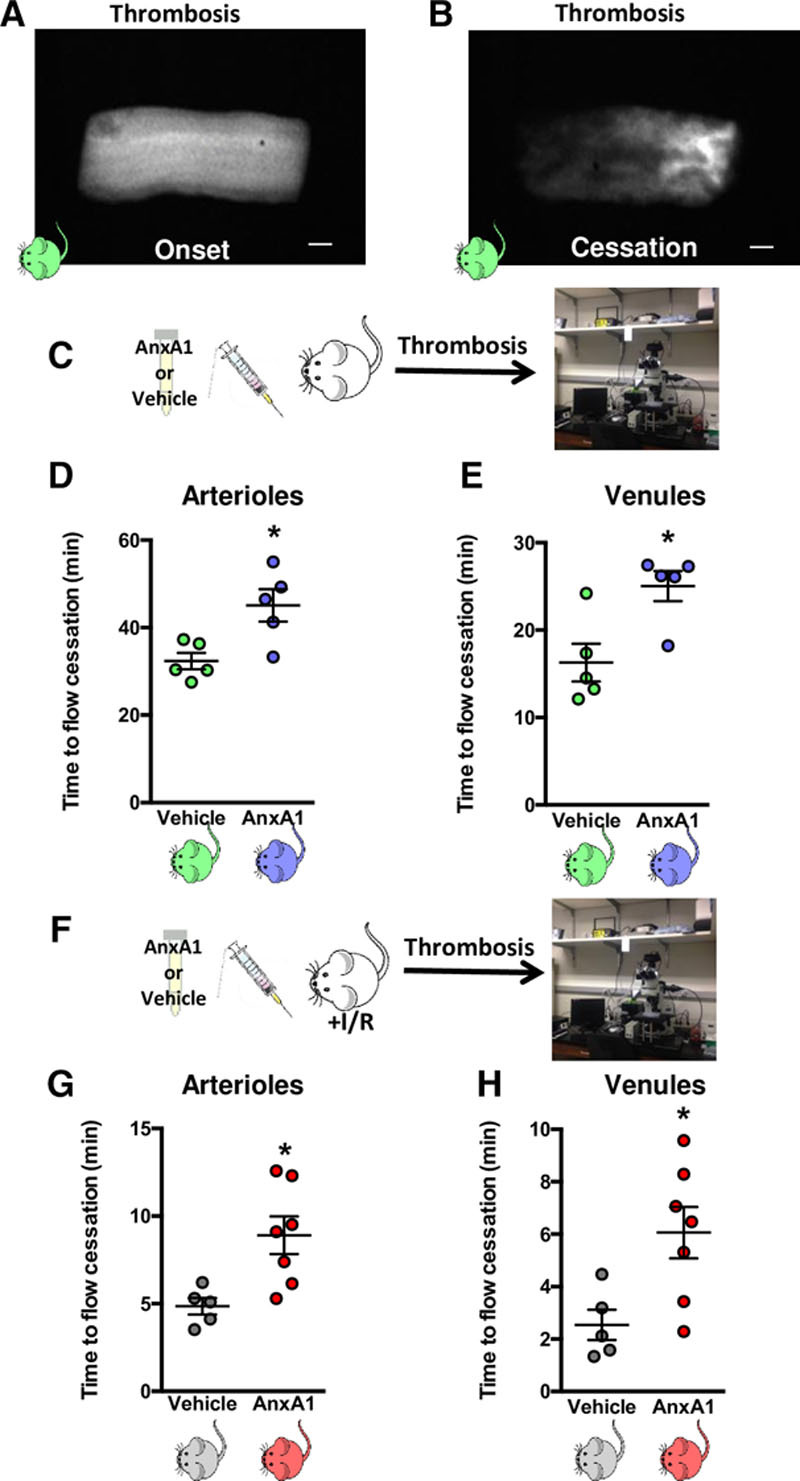
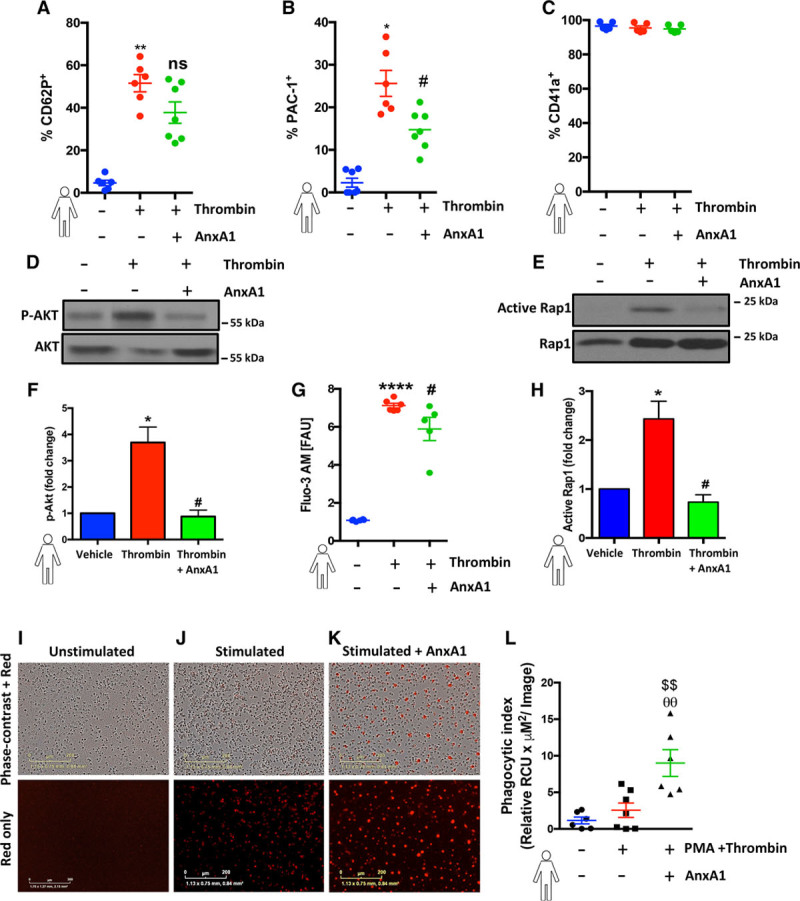
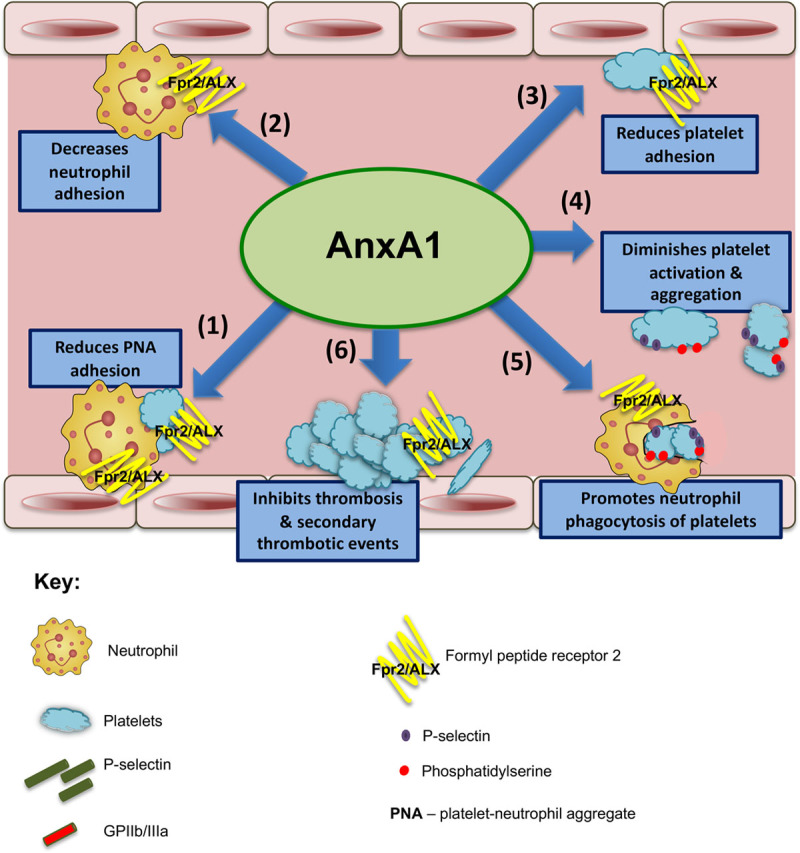
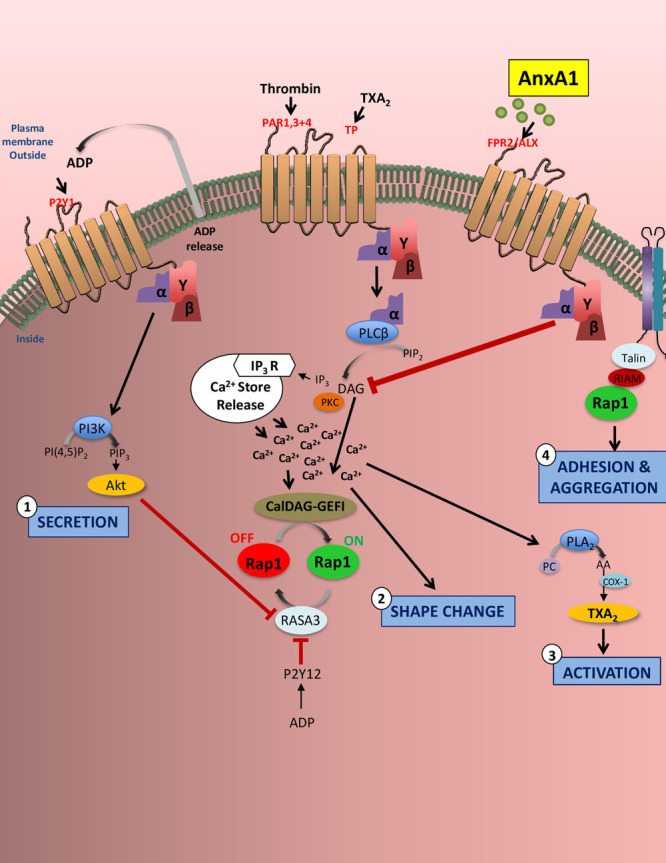
Results: Intravital microscopy revealed heightened platelet adherence and aggregate formation post I/RI, which were further exacerbated in AnxA1-/- mice. AnxA1 administration regulated platelet function directly (eg, via reducing thromboxane B2 and modulating phosphatidylserine expression) to promote cerebral protection post-I/RI and act as an effective preventative strategy for stroke by reducing platelet activation, aggregate formation, and cerebral thrombosis, a prerequisite for ischemic stroke. To translate these findings into a clinical setting, we show that AnxA1 plasma levels are reduced in human and murine stroke and that AnxA1 is able to act on human platelets, suppressing classic thrombin-induced inside-out signaling events (eg, Akt activation, intracellular calcium release, and Ras-associated protein 1 [Rap1] expression) to decrease αIIbβ3 activation without altering its surface expression. AnxA1 also selectively modifies cell surface determinants (eg, phosphatidylserine) to promote platelet phagocytosis by neutrophils, thereby driving active resolution. (n=5-13 mice/group or 7-10 humans/group.) Conclusions: AnxA1 affords protection by altering the platelet phenotype in cerebral I/RI from propathogenic to regulatory and reducing the propensity for platelets to aggregate and cause thrombosis by affecting integrin (αIIbβ3) activation, a previously unknown phenomenon. Thus, our data reveal a novel multifaceted role for AnxA1 to act both as a therapeutic and a prophylactic drug via its ability to promote endogenous proresolving, antithromboinflammatory circuits in cerebral I/RI. Collectively, these results further advance our knowledge and understanding in the field of platelet and resolution biology.
Keywords: annexin A1; formyl peptide receptor; inflammation; integrins; stroke; thrombosis.
Figures
References
-
- Esmon CT. Crosstalk between inflammation and thrombosis. Maturitas. 2008;61:122–131. doi: 10.1016/j.maturitas.2003.10.015. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
