

Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases
- PMID: 31156446
- PMCID: PMC6533591
- DOI: 10.3389/fphys.2019.00517
Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases
Abstract
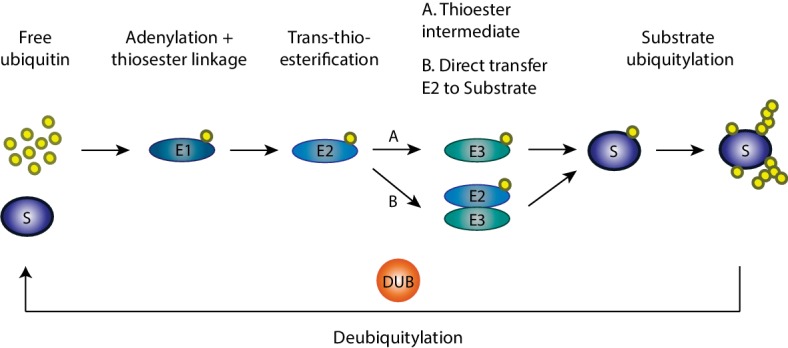
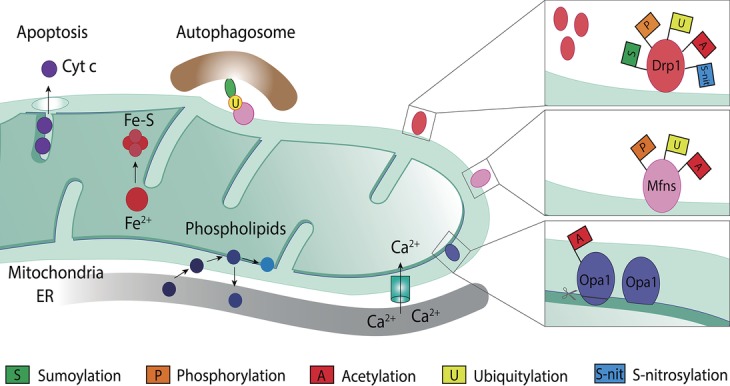
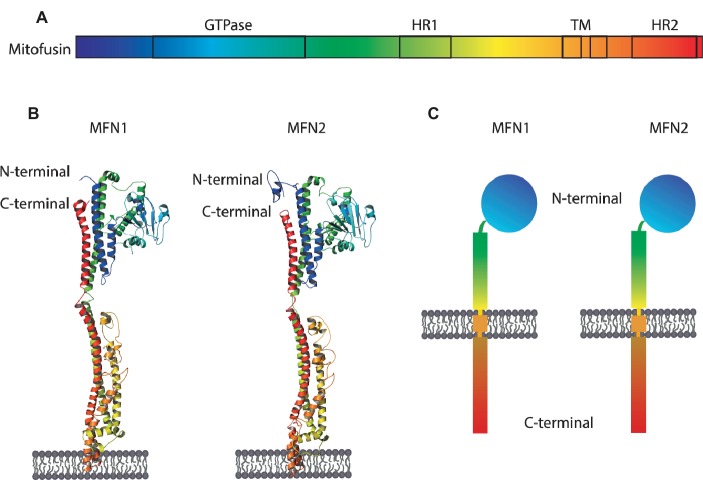
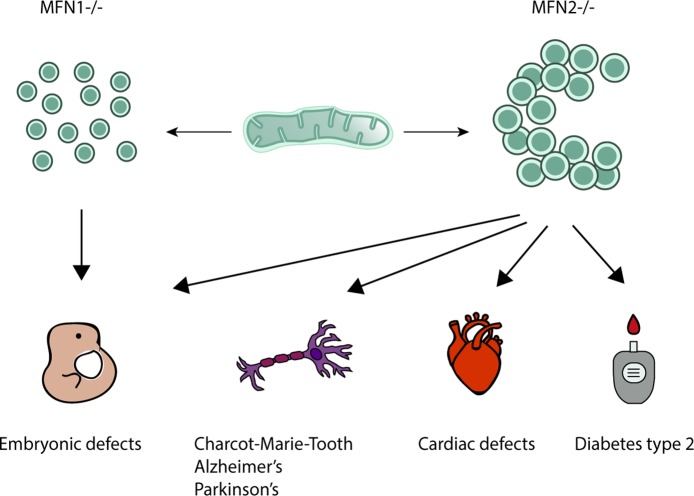
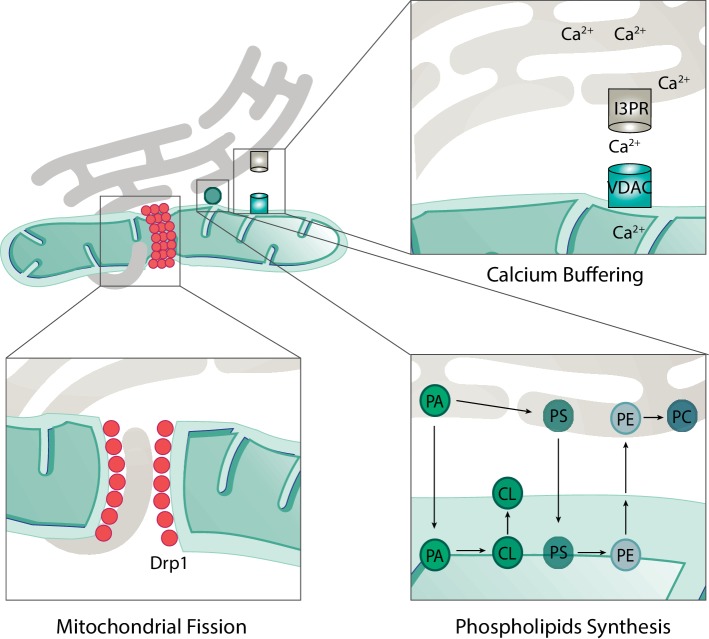
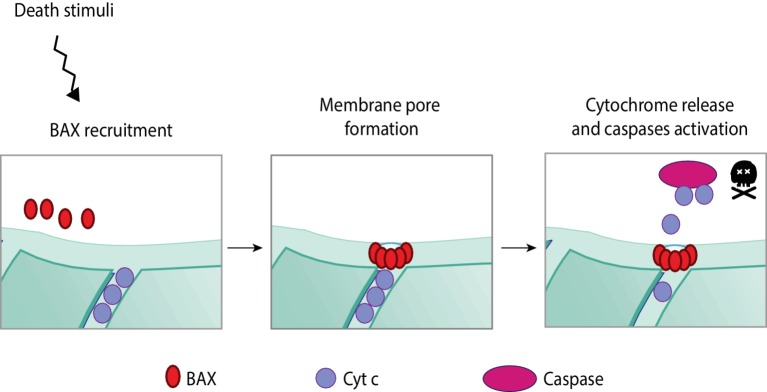
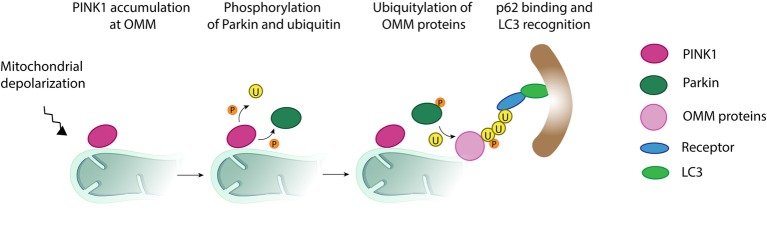
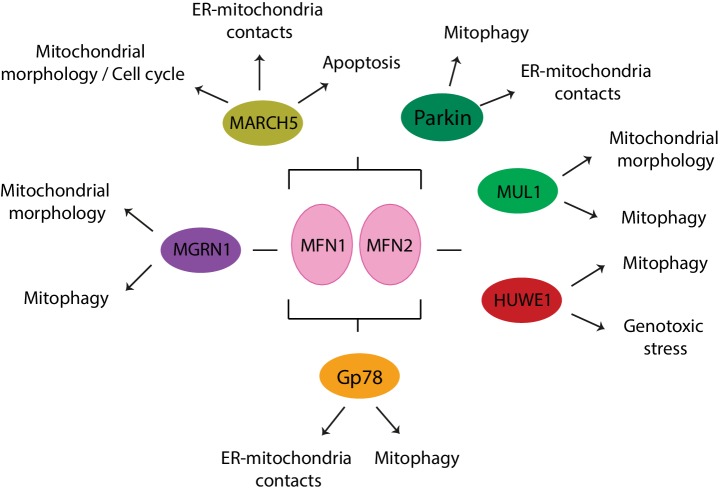
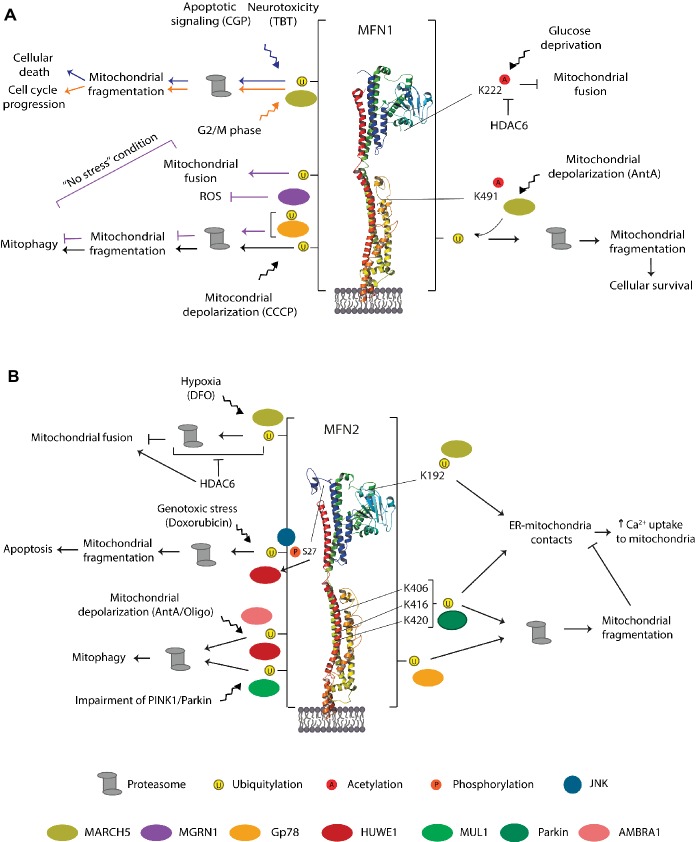
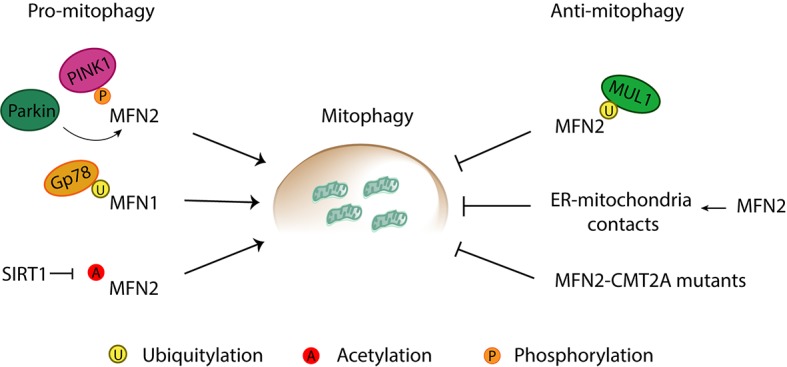
Mitochondria are dynamic organelles engaged in quality control and aging processes. They constantly undergo fusion, fission, transport, and anchoring events, which empower mitochondria with a very interactive behavior. The membrane remodeling processes needed for fusion require conserved proteins named mitofusins, MFN1 and MFN2 in mammals and Fzo1 in yeast. They are the first determinants deciding on whether communication and content exchange between different mitochondrial populations should occur. Importantly, each cell possesses hundreds of mitochondria, with a different severity of mitochondrial mutations or dysfunctional proteins, which potentially spread damage to the entire network. Therefore, the degree of their merging capacity critically influences cellular fitness. In turn, the mitochondrial network rapidly and dramatically changes in response to metabolic and environmental cues. Notably, cancer or obesity conditions, and stress experienced by neurons and cardiomyocytes, for example, triggers the downregulation of mitofusins and thus fragmentation of mitochondria. This places mitofusins upfront in sensing and transmitting stress. In fact, mitofusins are almost entirely exposed to the cytoplasm, a topology suitable for a critical relay point in information exchange between mitochondria and their cellular environment. Consistent with their topology, mitofusins are either activated or repressed by cytosolic post-translational modifiers, mainly by ubiquitin. Ubiquitin is a ubiquitous small protein orchestrating multiple quality control pathways, which is covalently attached to lysine residues in its substrates, or in ubiquitin itself. Importantly, from a chain of events also mediated by E1 and E2 enzymes, E3 ligases perform the ultimate and determinant step in substrate choice. Here, we review the ubiquitin E3 ligases that modify mitofusins. Two mitochondrial E3 enzymes-March5 and MUL1-one ligase located to the ER-Gp78-and finally three cytosolic enzymes-MGRN1, HUWE1, and Parkin-were shown to ubiquitylate mitofusins, in response to a variety of cellular inputs. The respective outcomes on mitochondrial morphology, on contact sites to the endoplasmic reticulum and on destructive processes, like mitophagy or apoptosis, are presented. Ultimately, understanding the mechanisms by which E3 ligases and mitofusins sense and bi-directionally signal mitochondria-cytosolic dysfunctions could pave the way for therapeutic approaches in neurodegenerative, cardiovascular, and obesity-linked diseases.
Keywords: E3 ligases; ER; MFN1/MFN2; mitochondria; mitofusins; mitophagy; quality control; ubiquitin.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous