

Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing
- PMID: 31159850
- PMCID: PMC6547561
- DOI: 10.1186/s13059-019-1720-5
Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing
Abstract
Background: Structural variations (SVs) or copy number variations (CNVs) greatly impact the functions of the genes encoded in the genome and are responsible for diverse human diseases. Although a number of existing SV detection algorithms can detect many types of SVs using whole genome sequencing (WGS) data, no single algorithm can call every type of SVs with high precision and high recall.
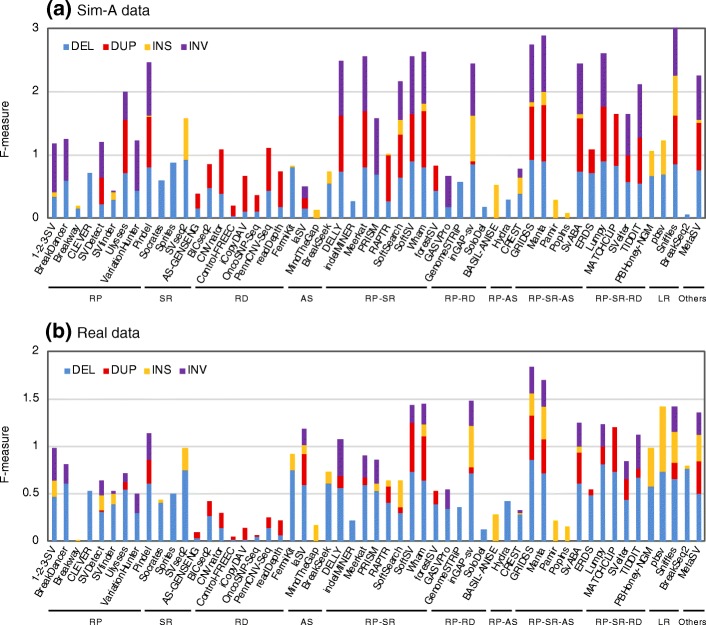
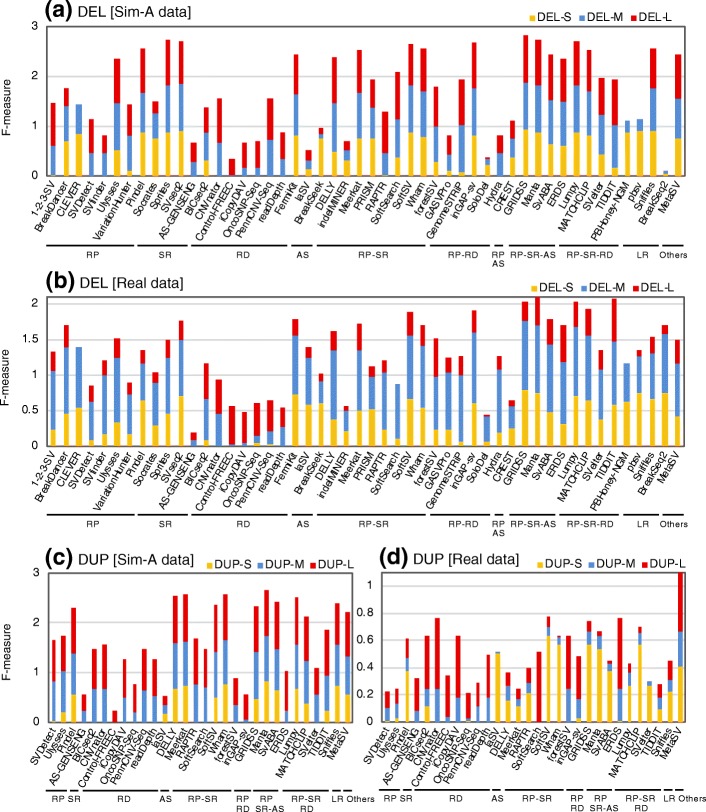
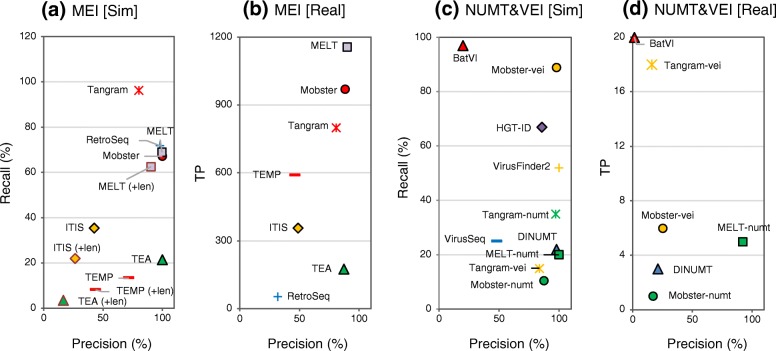
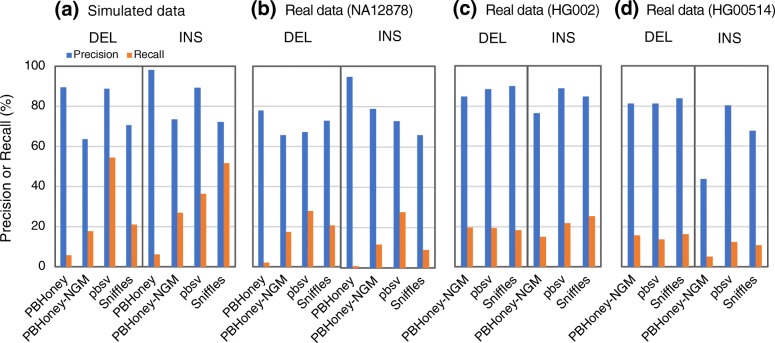
Results: We comprehensively evaluate the performance of 69 existing SV detection algorithms using multiple simulated and real WGS datasets. The results highlight a subset of algorithms that accurately call SVs depending on specific types and size ranges of the SVs and that accurately determine breakpoints, sizes, and genotypes of the SVs. We enumerate potential good algorithms for each SV category, among which GRIDSS, Lumpy, SVseq2, SoftSV, Manta, and Wham are better algorithms in deletion or duplication categories. To improve the accuracy of SV calling, we systematically evaluate the accuracy of overlapping calls between possible combinations of algorithms for every type and size range of SVs. The results demonstrate that both the precision and recall for overlapping calls vary depending on the combinations of specific algorithms rather than the combinations of methods used in the algorithms.
Conclusion: These results suggest that careful selection of the algorithms for each type and size range of SVs is required for accurate calling of SVs. The selection of specific pairs of algorithms for overlapping calls promises to effectively improve the SV detection accuracy.
Keywords: CNV; Copy number variation; Next generation sequencing; SV; Structural variation; WGS.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
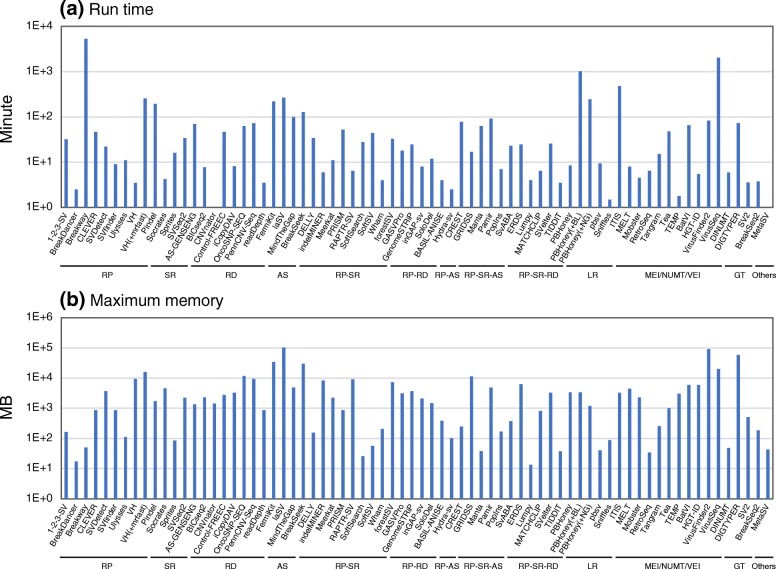
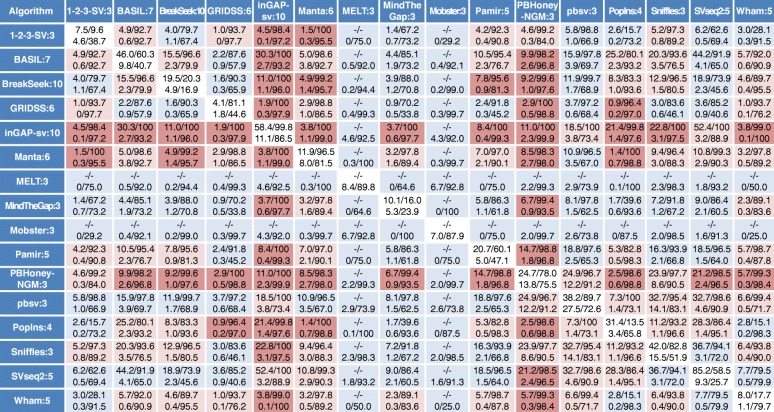
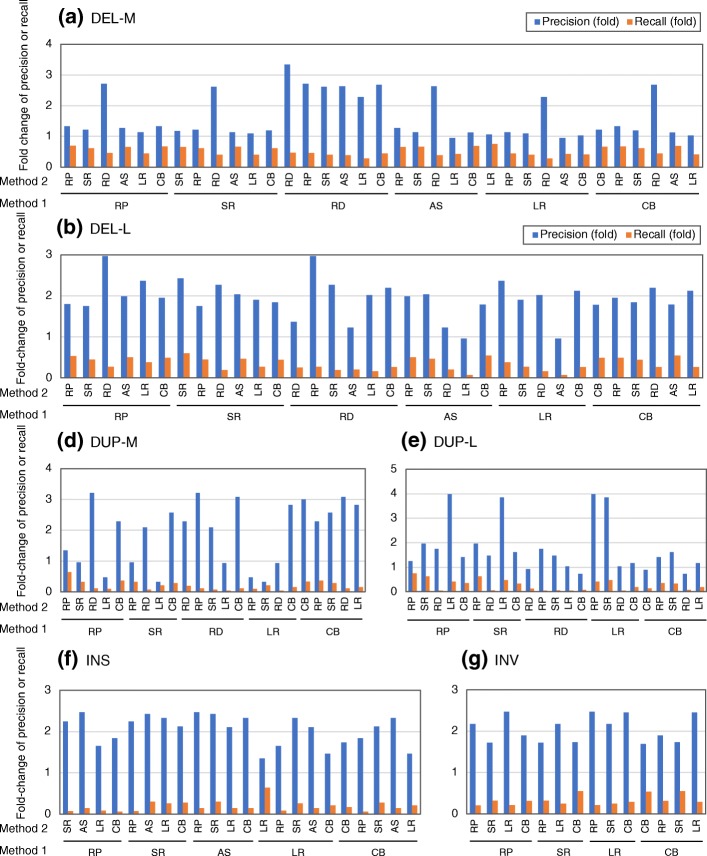
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
