

Haplotype-aware diplotyping from noisy long reads
- PMID: 31159868
- PMCID: PMC6547545
- DOI: 10.1186/s13059-019-1709-0
Haplotype-aware diplotyping from noisy long reads
Abstract
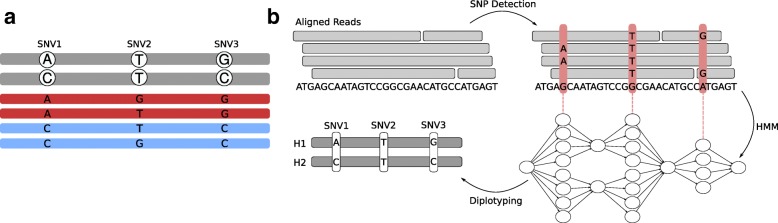
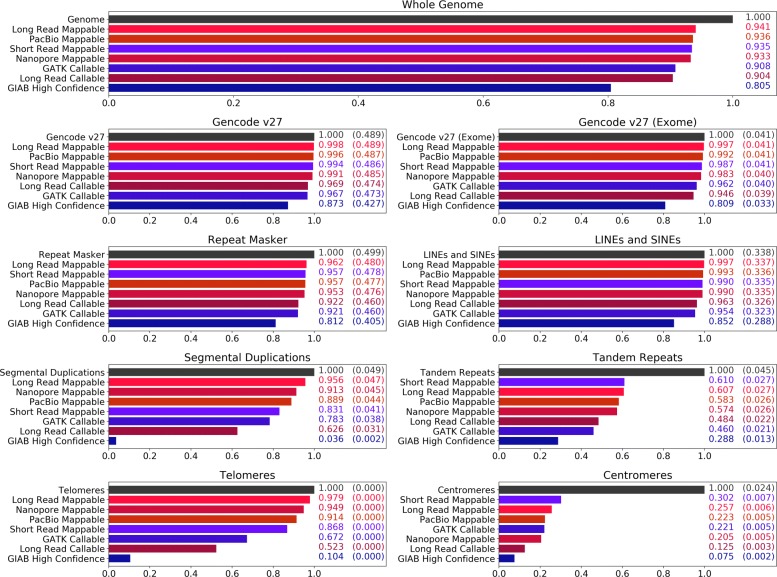
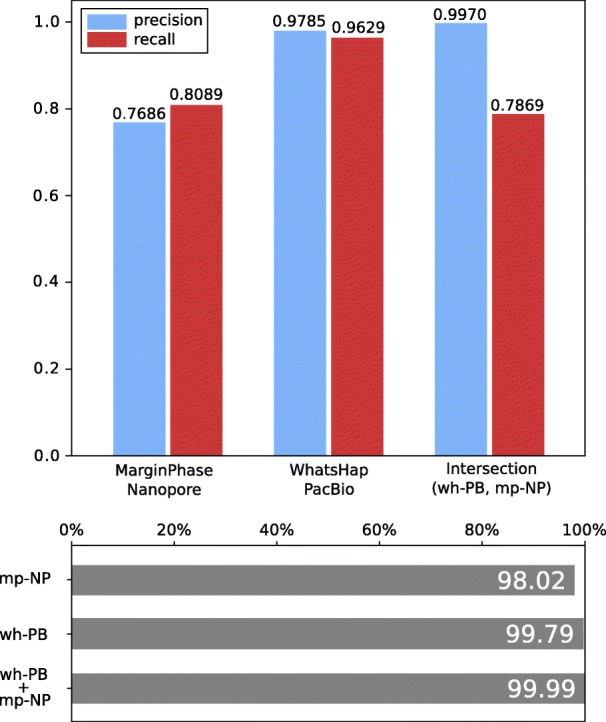
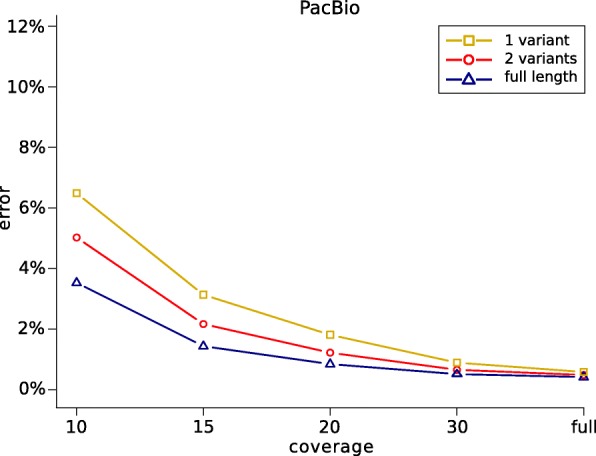
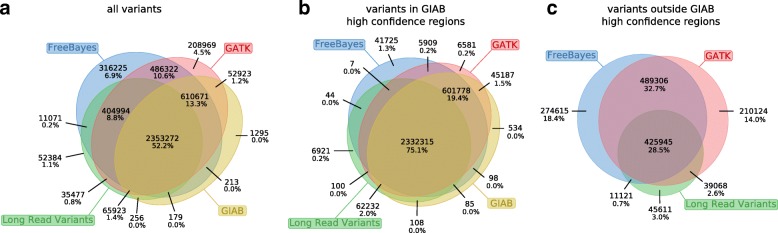
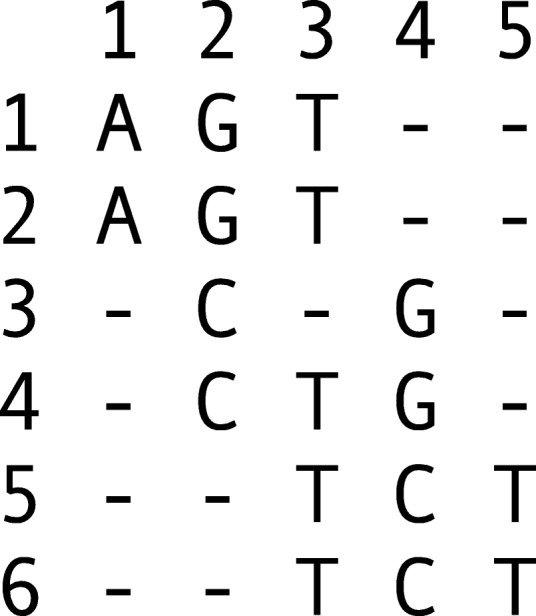
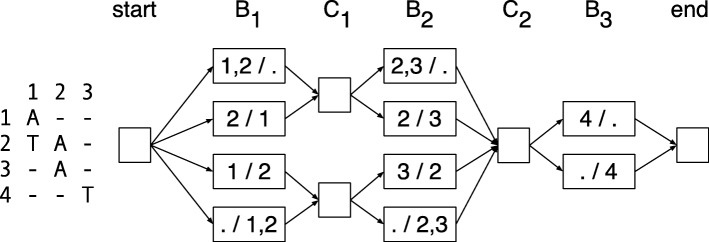
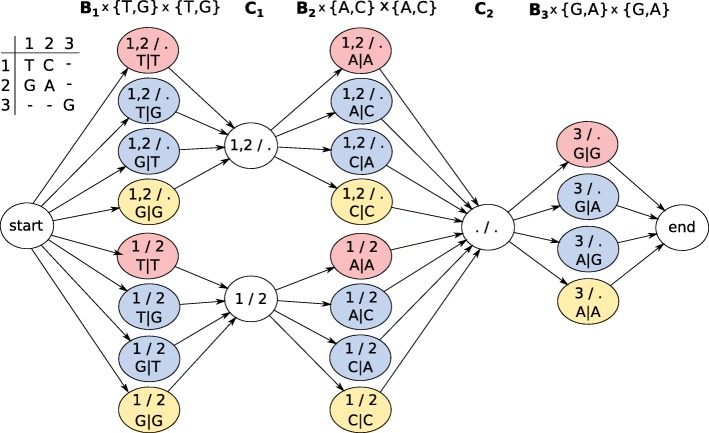
Current genotyping approaches for single-nucleotide variations rely on short, accurate reads from second-generation sequencing devices. Presently, third-generation sequencing platforms are rapidly becoming more widespread, yet approaches for leveraging their long but error-prone reads for genotyping are lacking. Here, we introduce a novel statistical framework for the joint inference of haplotypes and genotypes from noisy long reads, which we term diplotyping. Our technique takes full advantage of linkage information provided by long reads. We validate hundreds of thousands of candidate variants that have not yet been included in the high-confidence reference set of the Genome-in-a-Bottle effort.
Keywords: Computational genomics; Diplotypes; Genotyping; Haplotypes; Long reads; Phasing.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
