

Genetic diversity of CHC22 clathrin impacts its function in glucose metabolism
- PMID: 31159924
- PMCID: PMC6548504
- DOI: 10.7554/eLife.41517
Genetic diversity of CHC22 clathrin impacts its function in glucose metabolism
Abstract
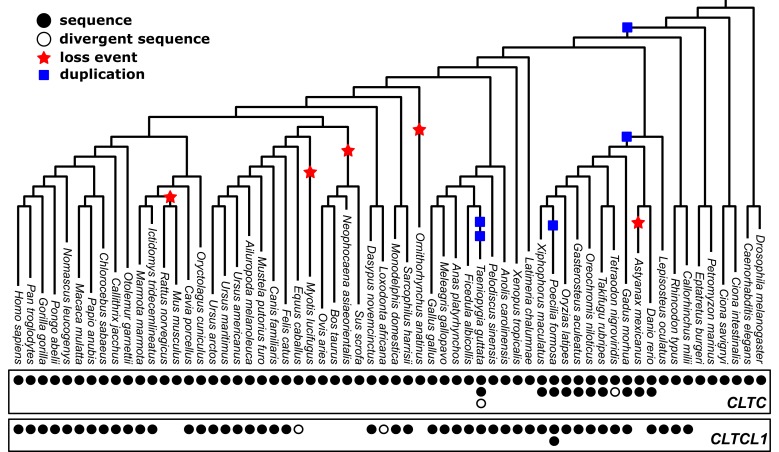
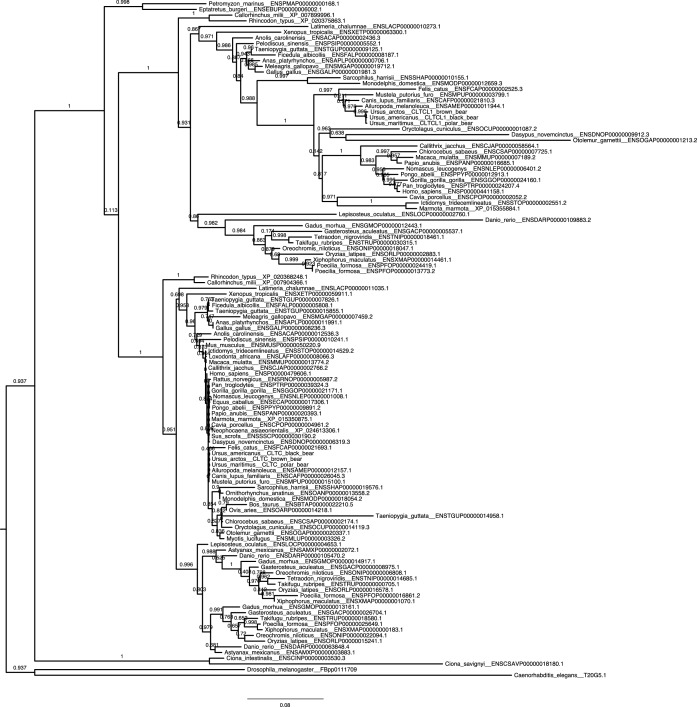
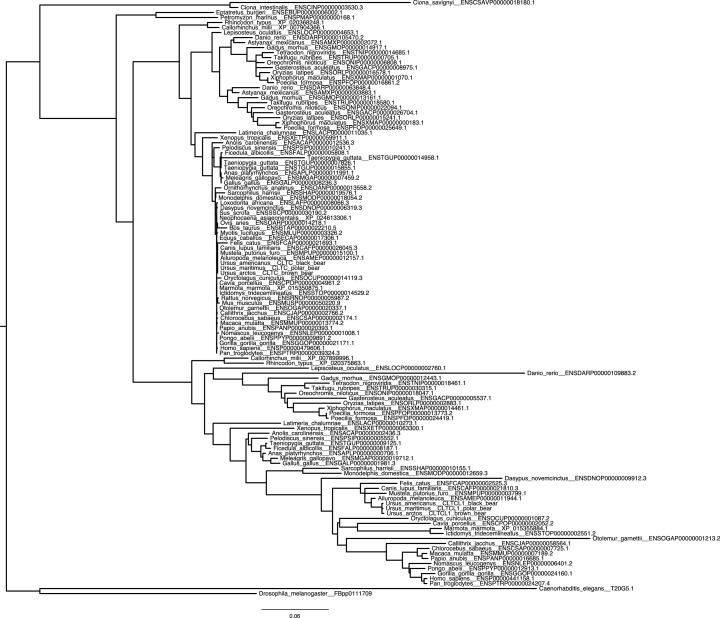
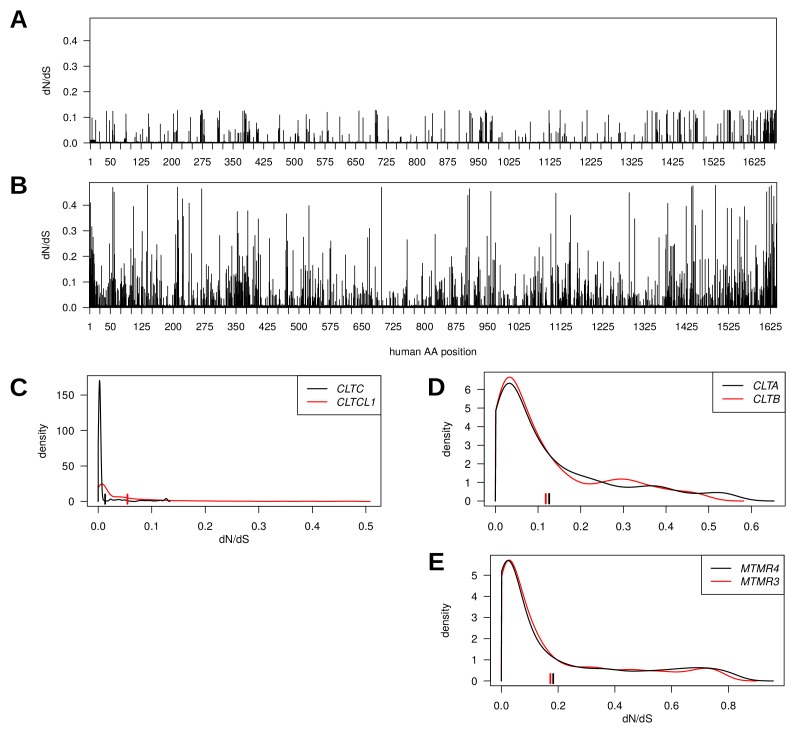
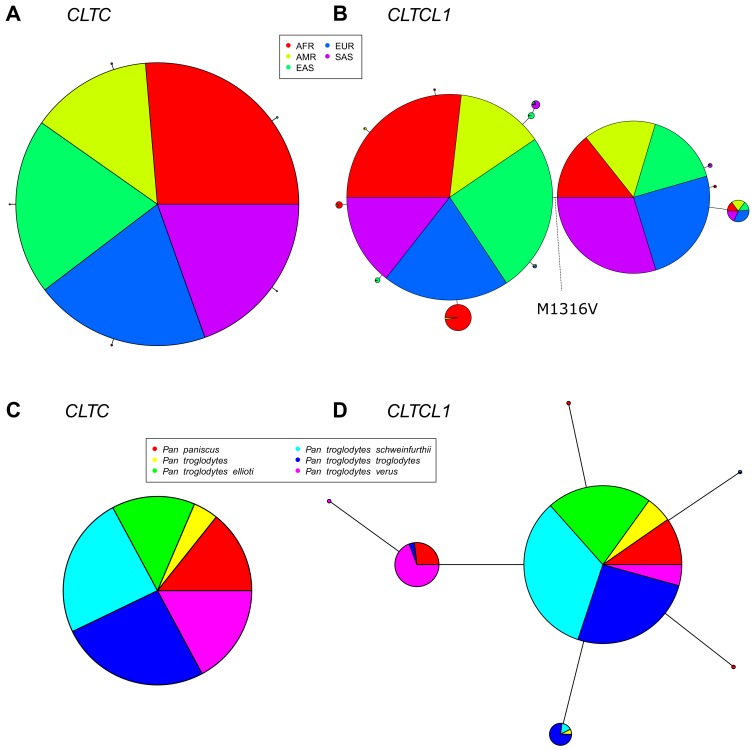
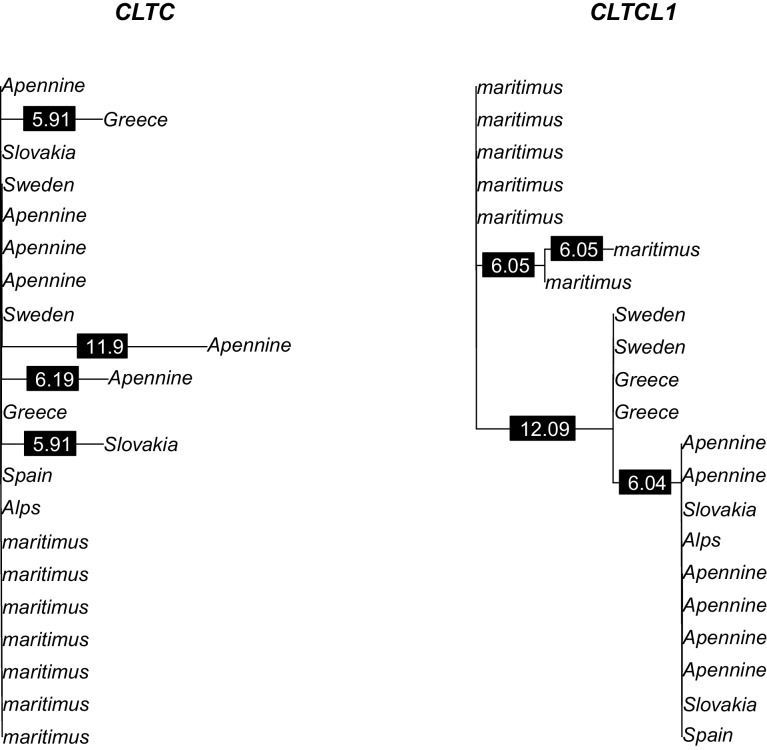
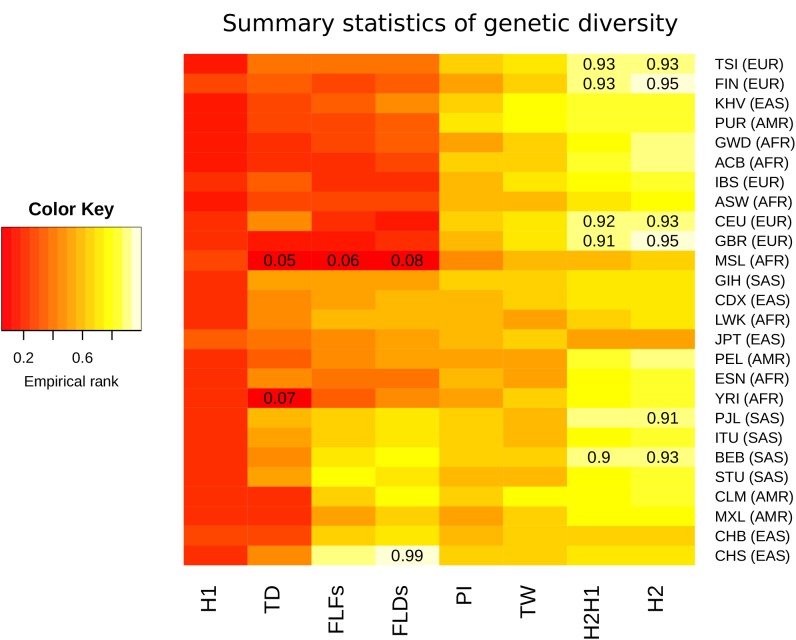
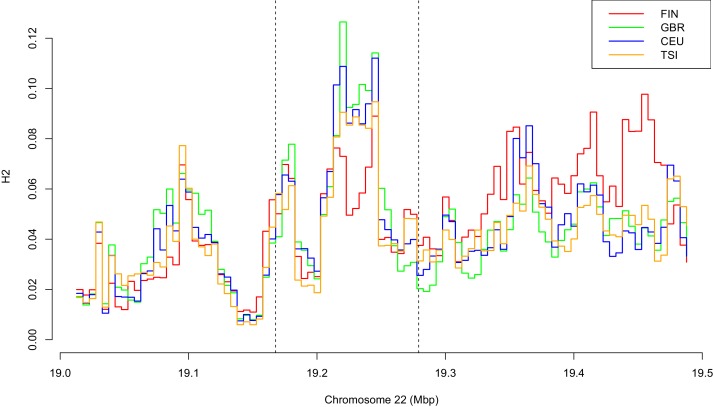
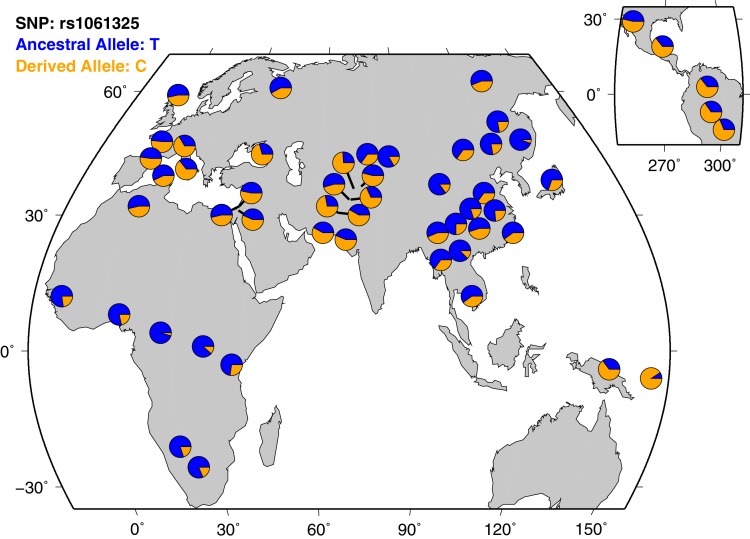
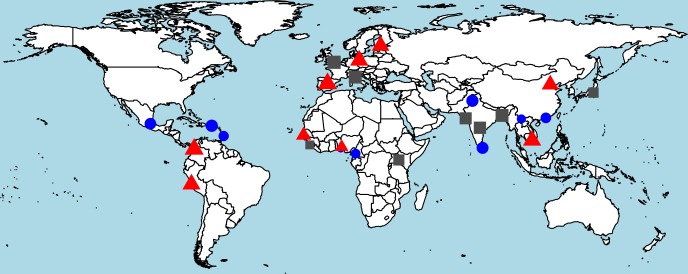
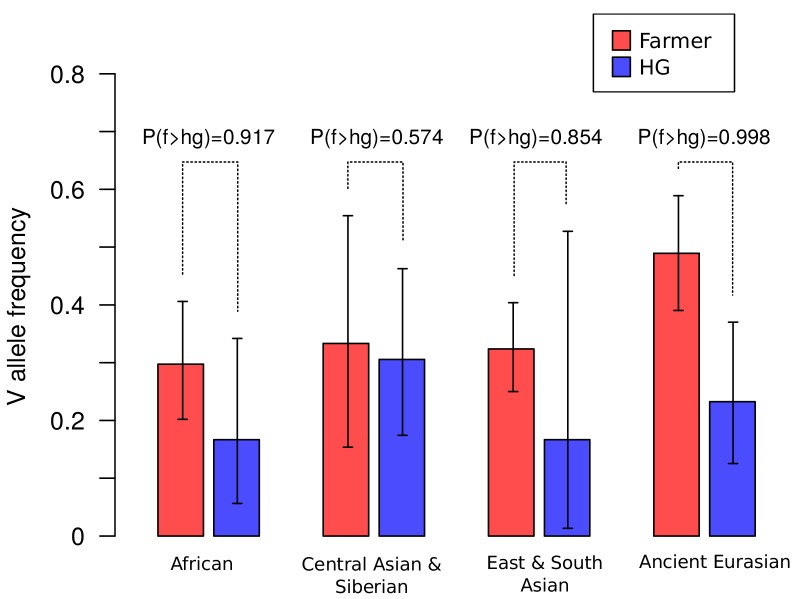
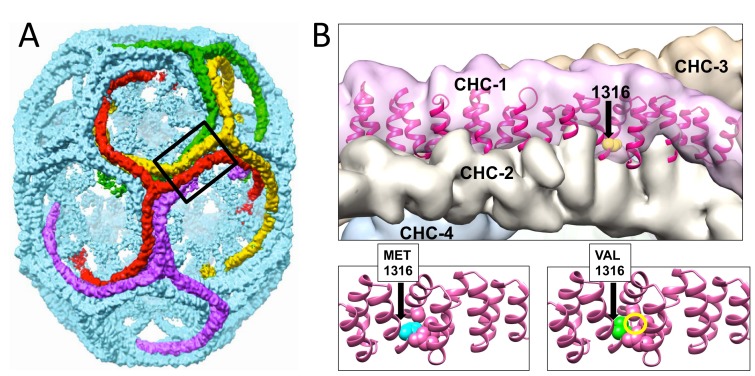
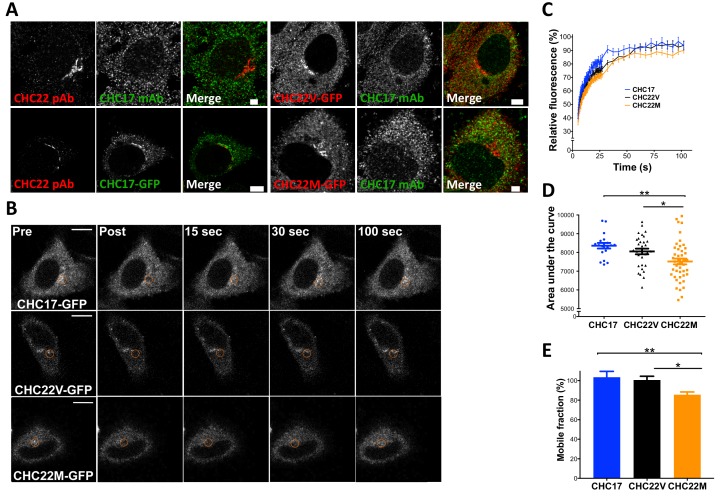
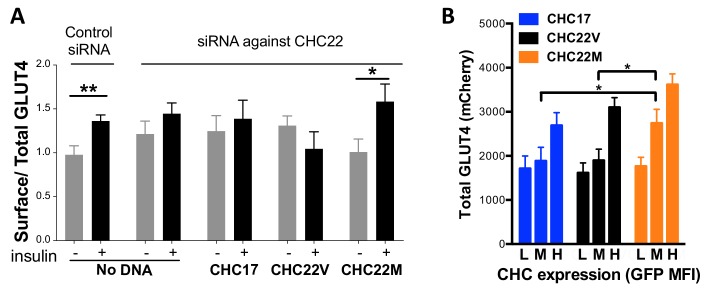
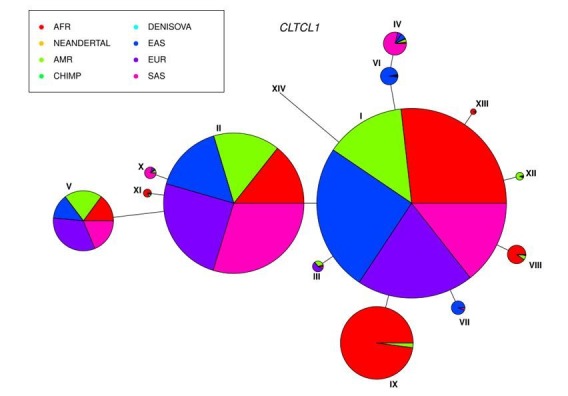
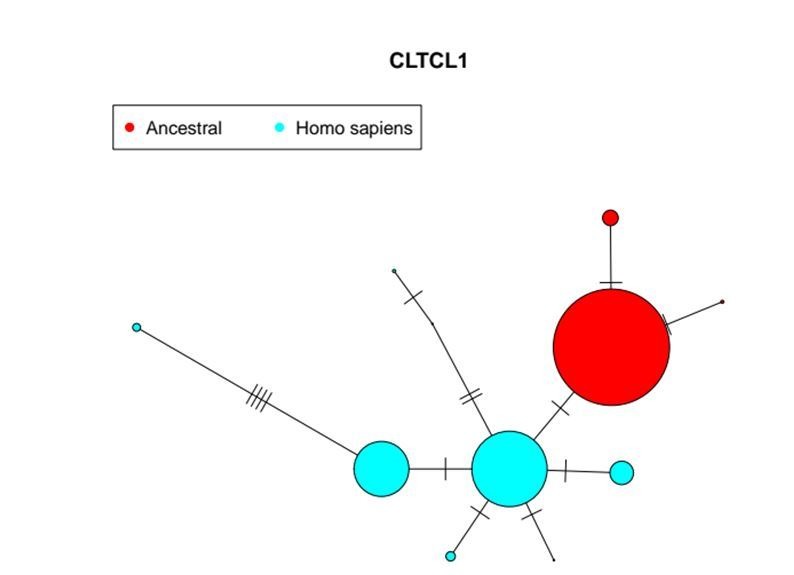
CHC22 clathrin plays a key role in intracellular membrane traffic of the insulin-responsive glucose transporter GLUT4 in humans. We performed population genetic and phylogenetic analyses of the CHC22-encoding CLTCL1 gene, revealing independent gene loss in at least two vertebrate lineages, after arising from gene duplication. All vertebrates retained the paralogous CLTC gene encoding CHC17 clathrin, which mediates endocytosis. For vertebrates retaining CLTCL1, strong evidence for purifying selection supports CHC22 functionality. All human populations maintained two high frequency CLTCL1 allelic variants, encoding either methionine or valine at position 1316. Functional studies indicated that CHC22-V1316, which is more frequent in farming populations than in hunter-gatherers, has different cellular dynamics than M1316-CHC22 and is less effective at controlling GLUT4 membrane traffic, altering its insulin-regulated response. These analyses suggest that ancestral human dietary change influenced selection of allotypes that affect CHC22's role in metabolism and have potential to differentially influence the human insulin response.
Keywords: cell biology; evolutionary biology; evolutionary selection; human; insulin response; membrane traffic.
© 2019, Fumagalli et al.
Conflict of interest statement
MF, SC, YD, AB, MC, PN, AJ, LA, AB, RR, IM, MT, PP, MT, FB No competing interests declared
Figures
Similar articles
-
CHC22 and CHC17 clathrins have distinct biochemical properties and display differential regulation and function.J Biol Chem. 2017 Dec 22;292(51):20834-20844. doi: 10.1074/jbc.M117.816256. Epub 2017 Nov 2. J Biol Chem. 2017. PMID: 29097553 Free PMC article.
-
Clathrin heavy and light chain isoforms originated by independent mechanisms of gene duplication during chordate evolution.Proc Natl Acad Sci U S A. 2005 May 17;102(20):7209-14. doi: 10.1073/pnas.0502058102. Epub 2005 May 9. Proc Natl Acad Sci U S A. 2005. PMID: 15883369 Free PMC article.
-
The clathrin heavy chain isoform CHC22 functions in a novel endosomal sorting step.J Cell Biol. 2010 Jan 11;188(1):131-44. doi: 10.1083/jcb.200908057. J Cell Biol. 2010. PMID: 20065094 Free PMC article.
-
Building GLUT4 Vesicles: CHC22 Clathrin's Human Touch.Trends Cell Biol. 2020 Sep;30(9):705-719. doi: 10.1016/j.tcb.2020.05.007. Epub 2020 Jun 30. Trends Cell Biol. 2020. PMID: 32620516 Review.
-
Diversity of clathrin function: new tricks for an old protein.Annu Rev Cell Dev Biol. 2012;28:309-36. doi: 10.1146/annurev-cellbio-101011-155716. Epub 2012 Jul 23. Annu Rev Cell Dev Biol. 2012. PMID: 22831640 Review.
Cited by
-
WEIRD bodies: mismatch, medicine and missing diversity.Evol Hum Behav. 2020 Sep;41(5):330-340. doi: 10.1016/j.evolhumbehav.2020.04.001. Epub 2020 Apr 14. Evol Hum Behav. 2020. PMID: 33100820 Free PMC article.
-
Mechanistic divergences of endocytic clathrin-coated vesicle formation in mammals, yeasts and plants.J Cell Sci. 2024 Aug 15;137(16):jcs261847. doi: 10.1242/jcs.261847. Epub 2024 Aug 20. J Cell Sci. 2024. PMID: 39161994 Free PMC article. Review.
-
Food taboos during pregnancy: meta-analysis on cross cultural differences suggests specific, diet-related pressures on childbirth among agriculturalists.PeerJ. 2022 Jul 11;10:e13633. doi: 10.7717/peerj.13633. eCollection 2022. PeerJ. 2022. PMID: 35846875 Free PMC article.
-
Lipid Metabolism Links Nutrient-Exercise Timing to Insulin Sensitivity in Men Classified as Overweight or Obese.J Clin Endocrinol Metab. 2020 Mar 1;105(3):660-76. doi: 10.1210/clinem/dgz104. J Clin Endocrinol Metab. 2020. PMID: 31628477 Free PMC article. Clinical Trial.
-
The Potential Health Benefits of the Ketogenic Diet: A Narrative Review.Nutrients. 2021 May 13;13(5):1654. doi: 10.3390/nu13051654. Nutrients. 2021. PMID: 34068325 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
