

Single particle cryo-EM reconstruction of 52 kDa streptavidin at 3.2 Angstrom resolution
- PMID: 31160591
- PMCID: PMC6546690
- DOI: 10.1038/s41467-019-10368-w
Single particle cryo-EM reconstruction of 52 kDa streptavidin at 3.2 Angstrom resolution
Abstract
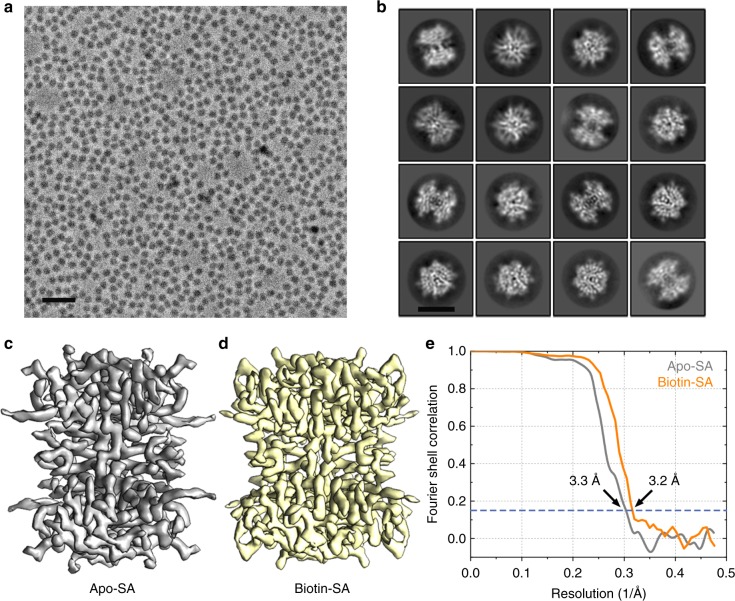
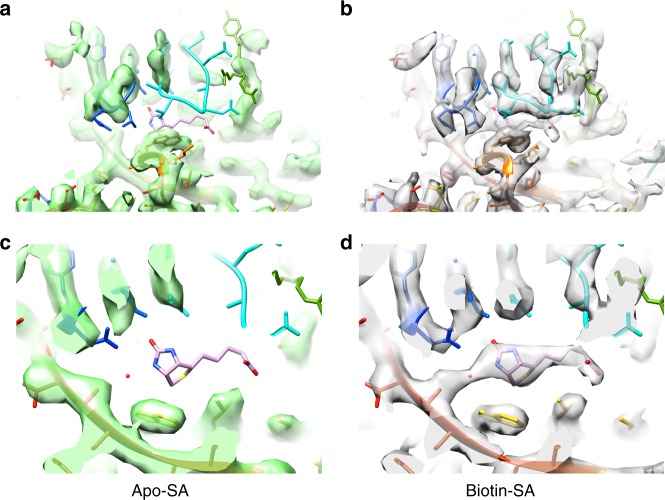
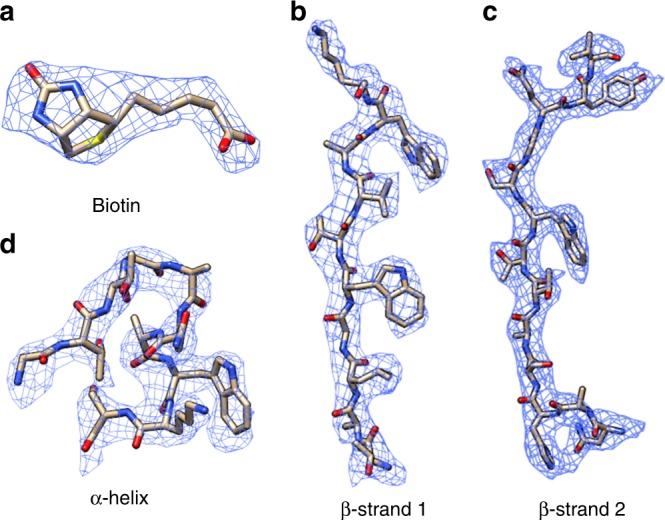
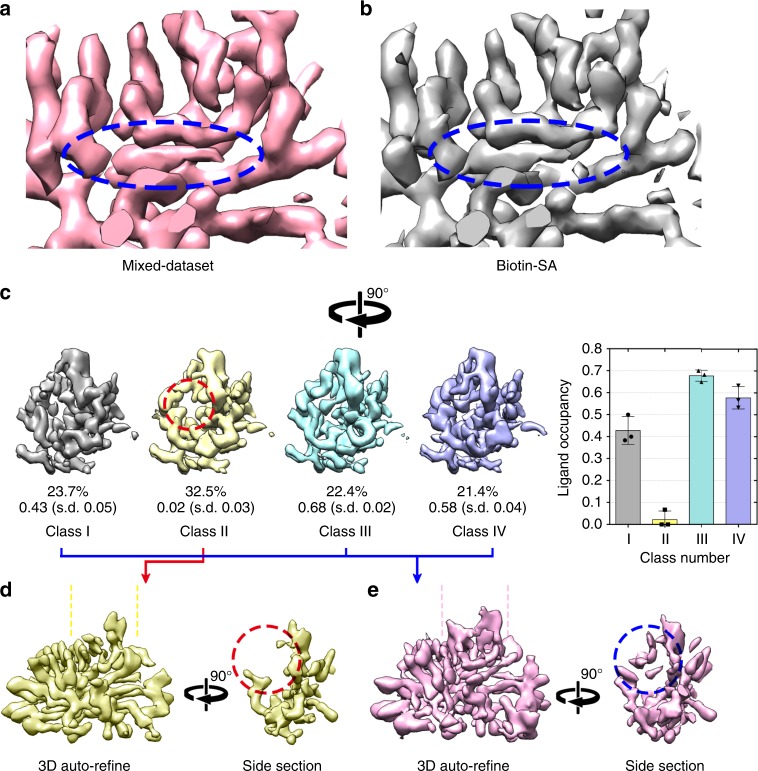
The fast development of single-particle cryogenic electron microscopy (cryo-EM) has made it more feasible to obtain the 3D structure of well-behaved macromolecules with a molecular weight higher than 300 kDa at ~3 Å resolution. However, it remains a challenge to obtain the high-resolution structures of molecules smaller than 200 kDa using single-particle cryo-EM. In this work, we apply the Cs-corrector-VPP-coupled cryo-EM to study the 52 kDa streptavidin (SA) protein supported on a thin layer of graphene and embedded in vitreous ice. We are able to solve both the apo-SA and biotin-bound SA structures at near-atomic resolution using single-particle cryo-EM. We demonstrate that the method has the potential to determine the structures of molecules as small as 39 kDa.
Conflict of interest statement
The authors declare no competing interests.
Figures
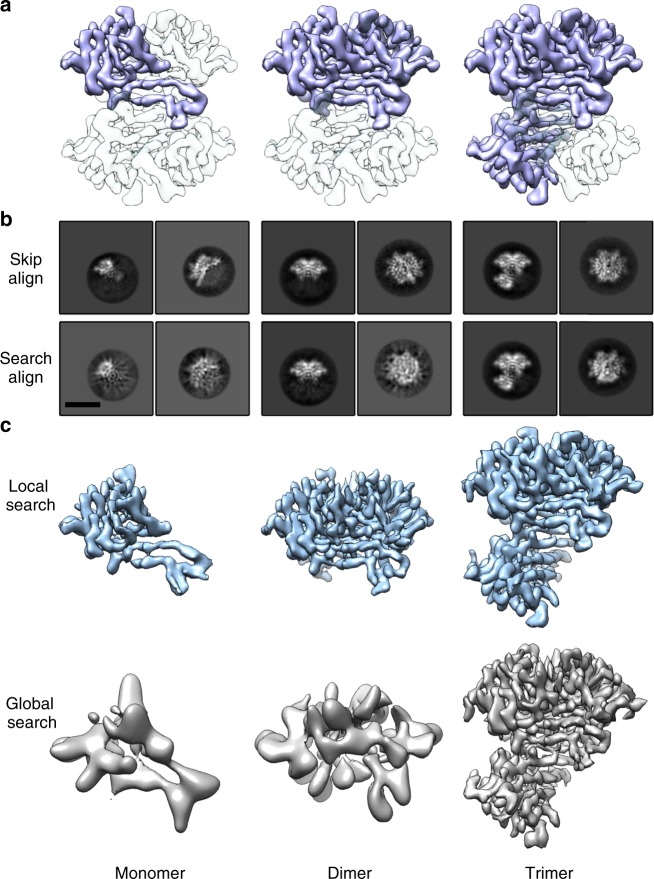
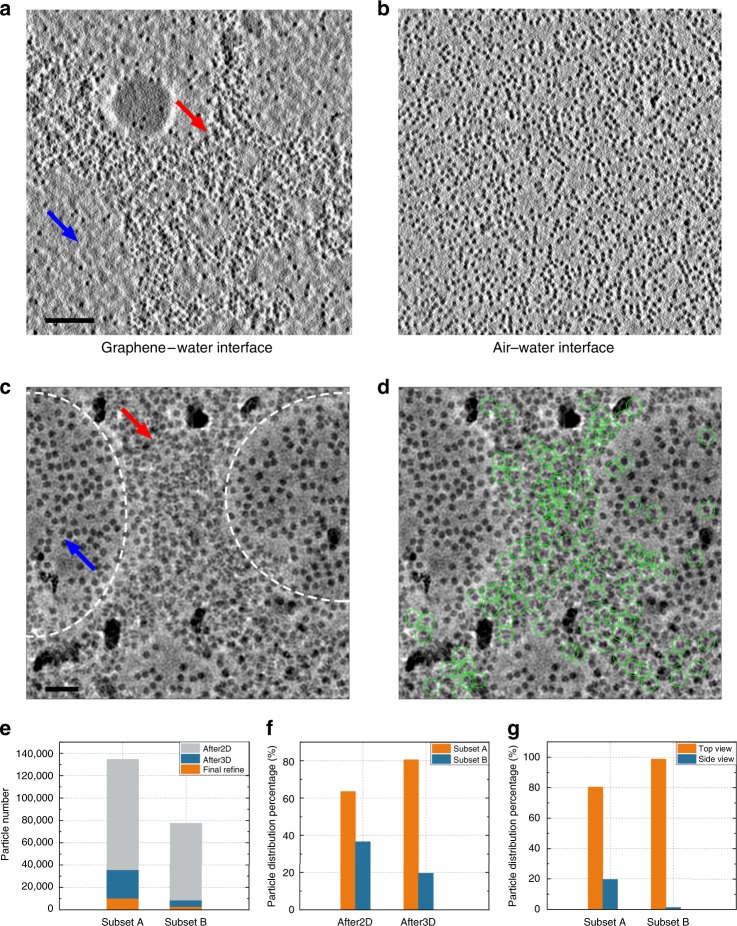
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
