

Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts
- PMID: 31160820
- PMCID: PMC6634302
- DOI: 10.1038/s41591-019-0457-8
Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts
Abstract
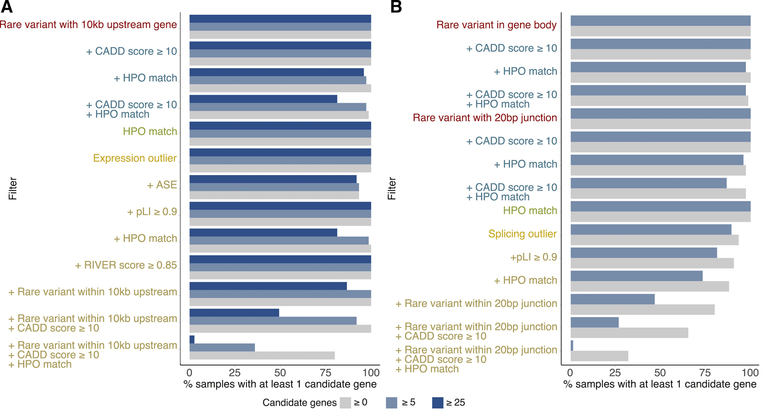
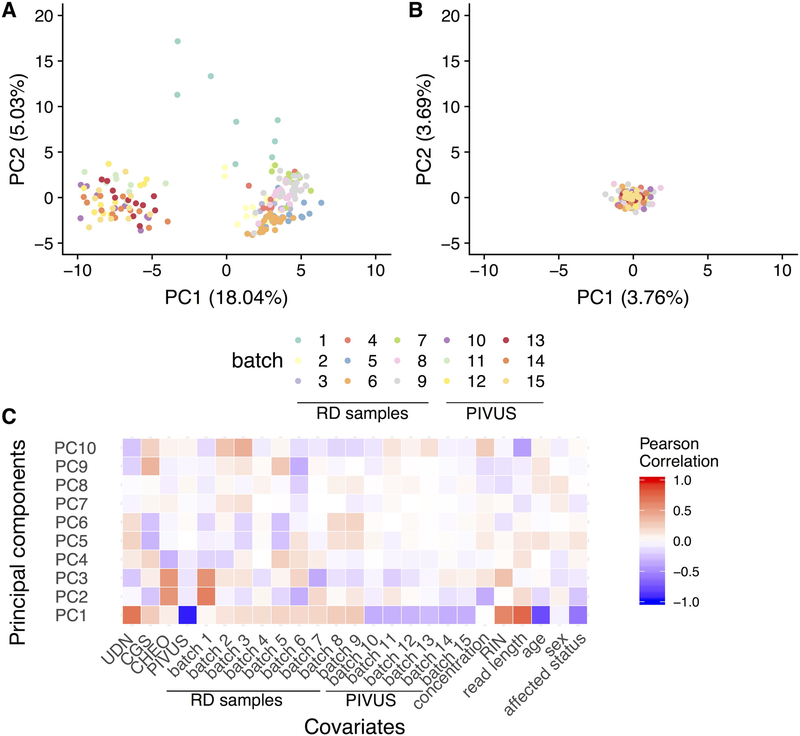
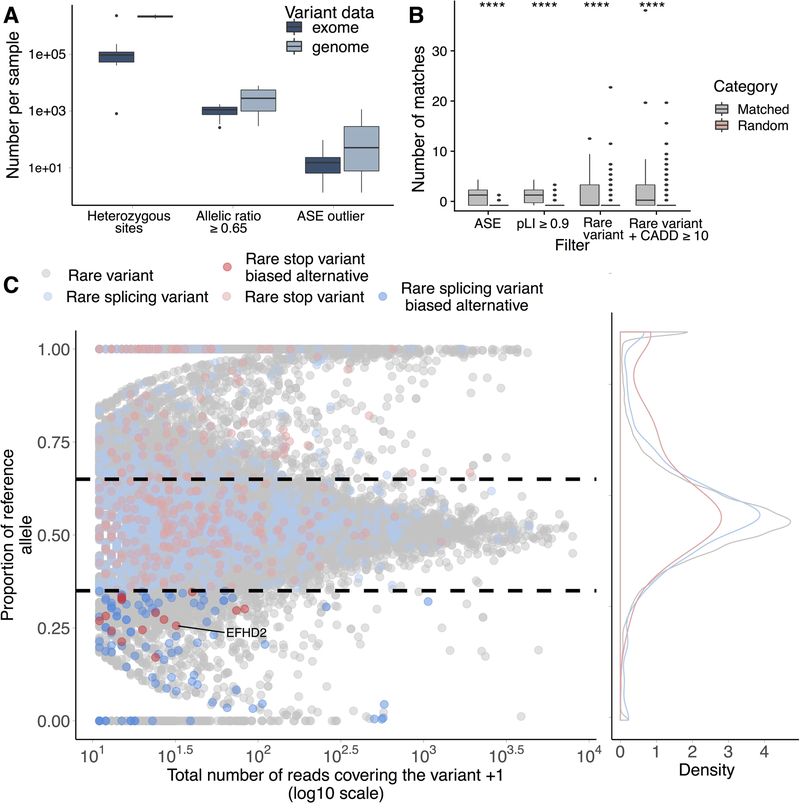
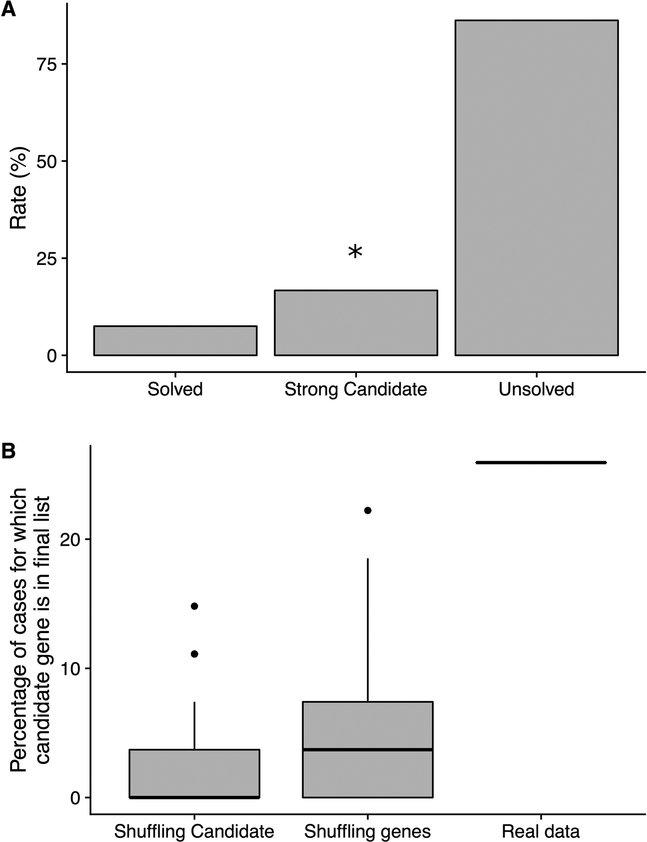
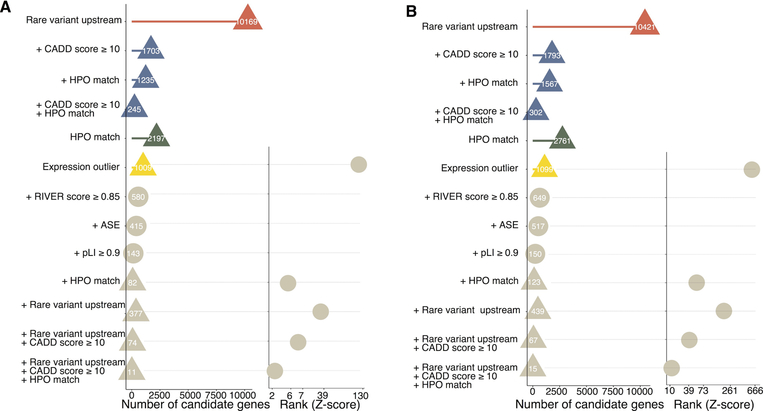
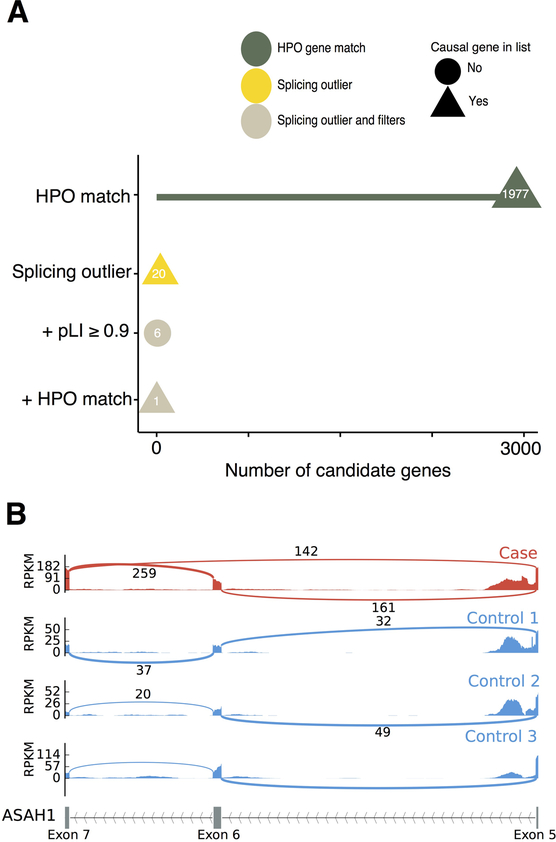
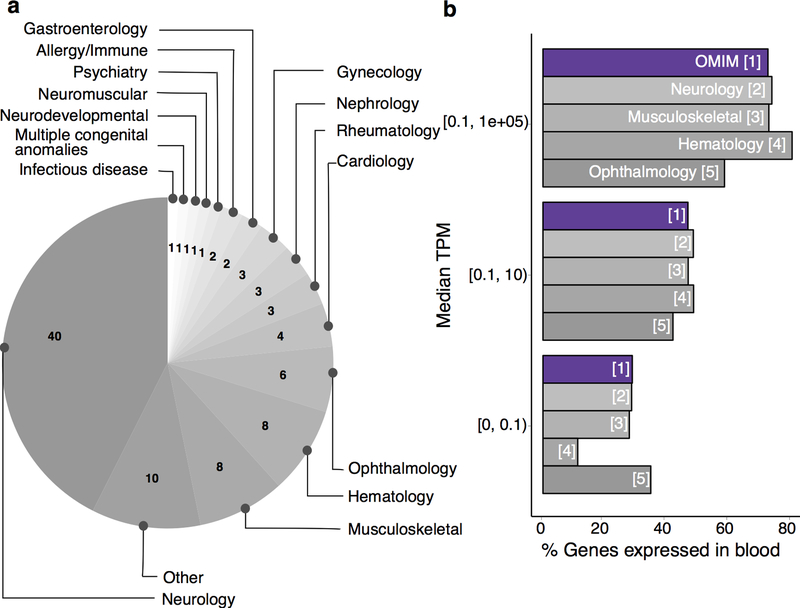
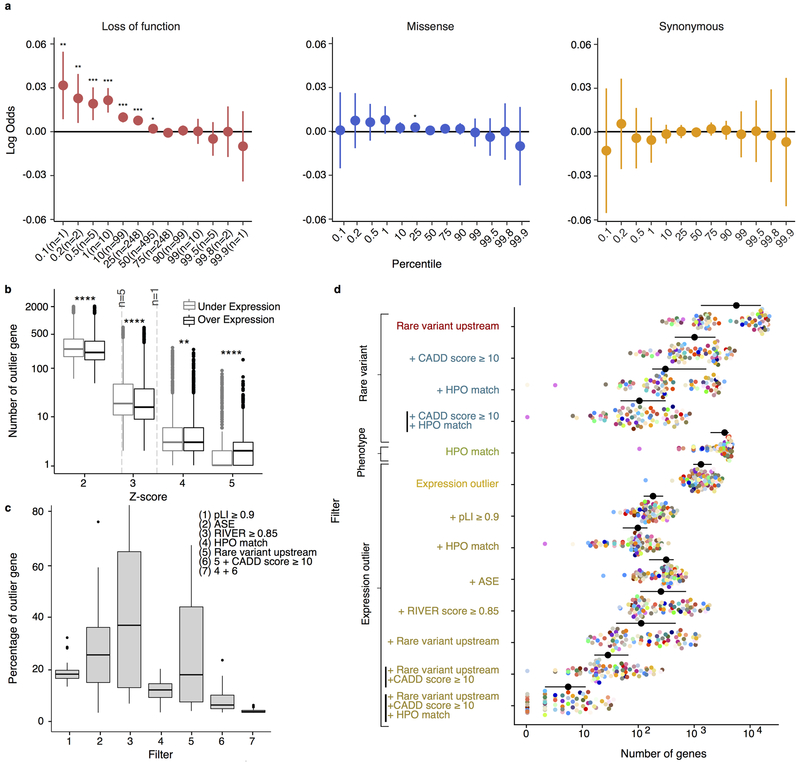
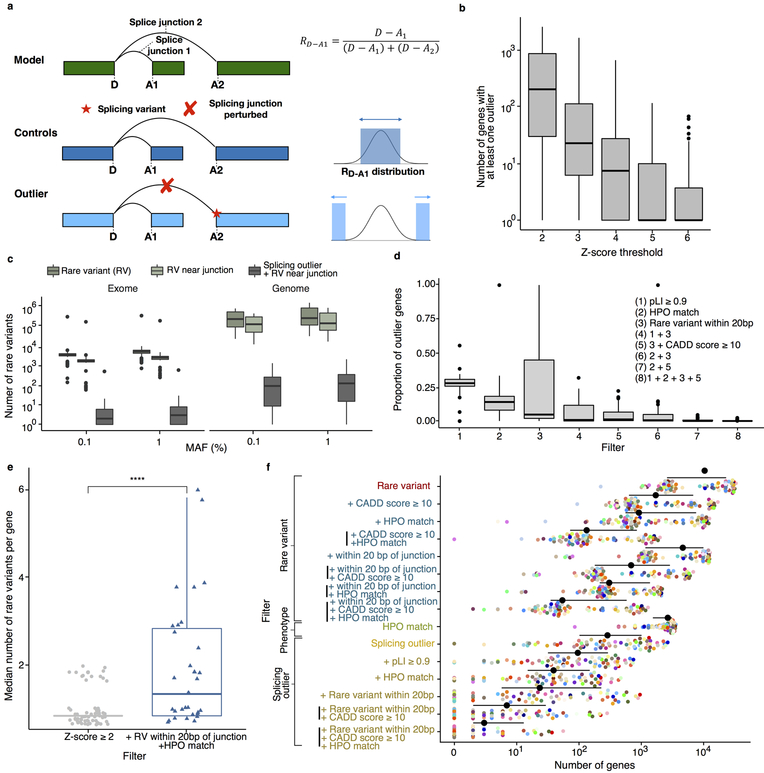
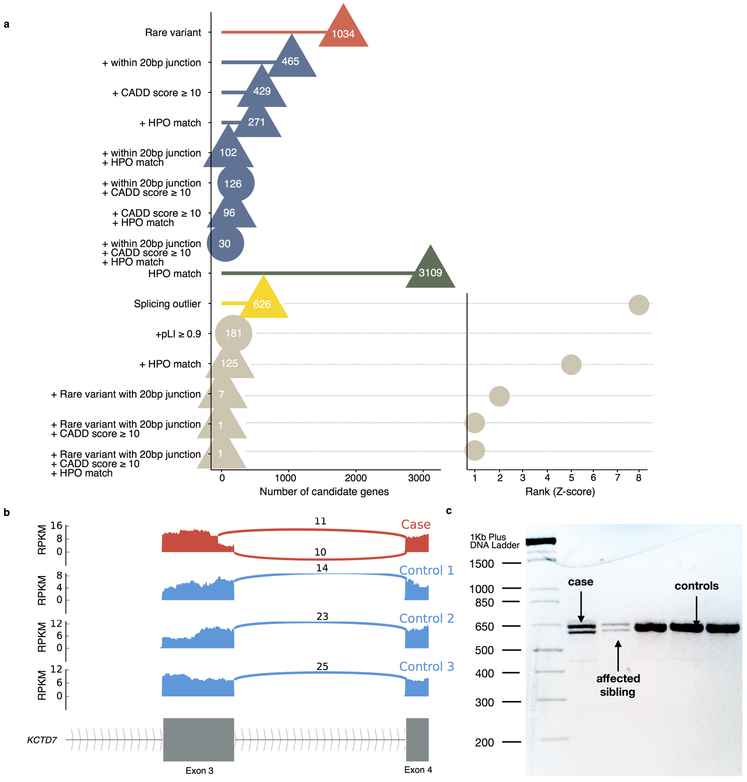
It is estimated that 350 million individuals worldwide suffer from rare diseases, which are predominantly caused by mutation in a single gene1. The current molecular diagnostic rate is estimated at 50%, with whole-exome sequencing (WES) among the most successful approaches2-5. For patients in whom WES is uninformative, RNA sequencing (RNA-seq) has shown diagnostic utility in specific tissues and diseases6-8. This includes muscle biopsies from patients with undiagnosed rare muscle disorders6,9, and cultured fibroblasts from patients with mitochondrial disorders7. However, for many individuals, biopsies are not performed for clinical care, and tissues are difficult to access. We sought to assess the utility of RNA-seq from blood as a diagnostic tool for rare diseases of different pathophysiologies. We generated whole-blood RNA-seq from 94 individuals with undiagnosed rare diseases spanning 16 diverse disease categories. We developed a robust approach to compare data from these individuals with large sets of RNA-seq data for controls (n = 1,594 unrelated controls and n = 49 family members) and demonstrated the impacts of expression, splicing, gene and variant filtering strategies on disease gene identification. Across our cohort, we observed that RNA-seq yields a 7.5% diagnostic rate, and an additional 16.7% with improved candidate gene resolution.
Conflict of interest statement
Competing Interests Statement
J.D.M. is on Genoox Scientific Advisory Board and Rainbow Genomics Clinical Advisory Board and consults for Illumina. E.A.A. is Co-Founder to Personalis, DeepCell and Advisor to Genome Medical and Sequence Bio. E.I. is a scientific advisor for Precision Wellness for work unrelated to the present project. S.B.M. is on the Scientific Advisory Board for Prime Genomics.
Figures
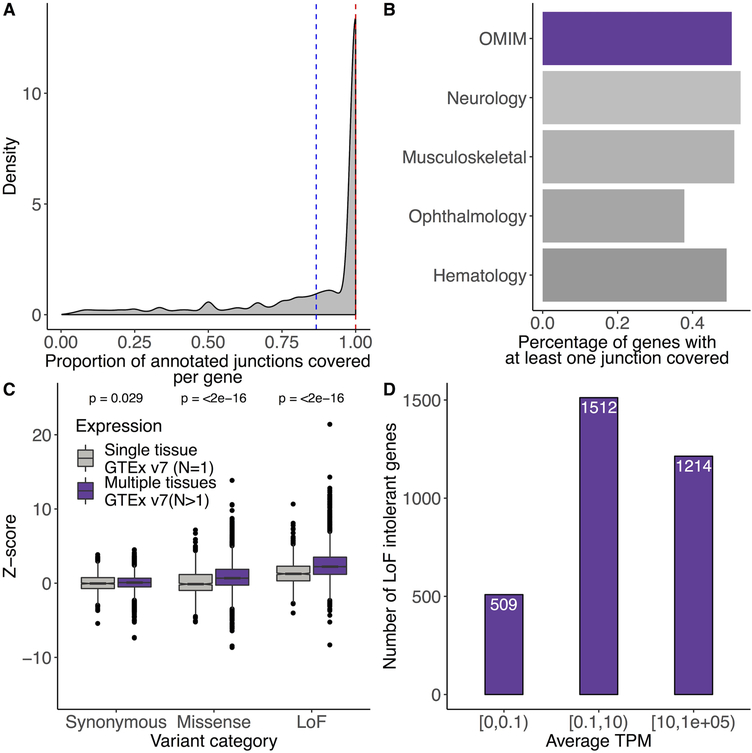
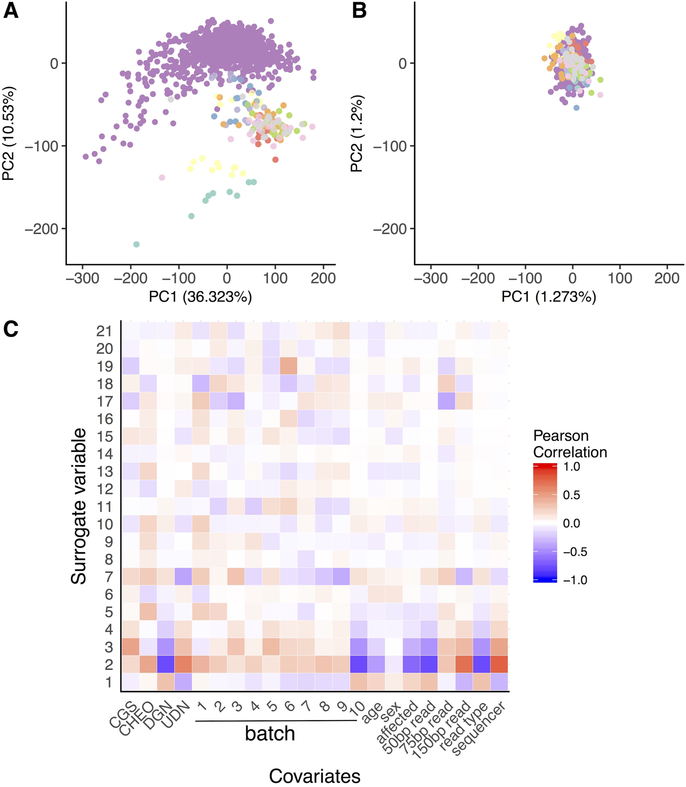
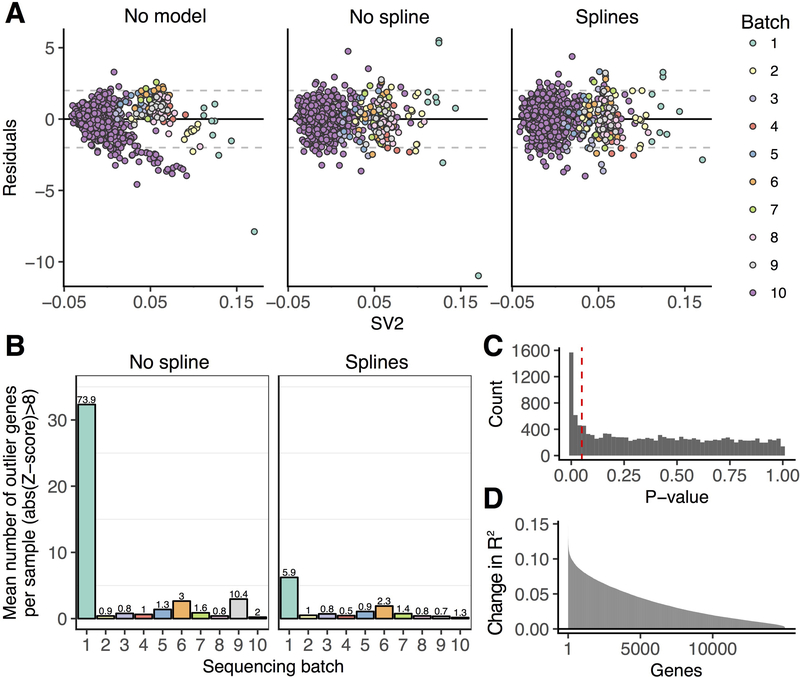
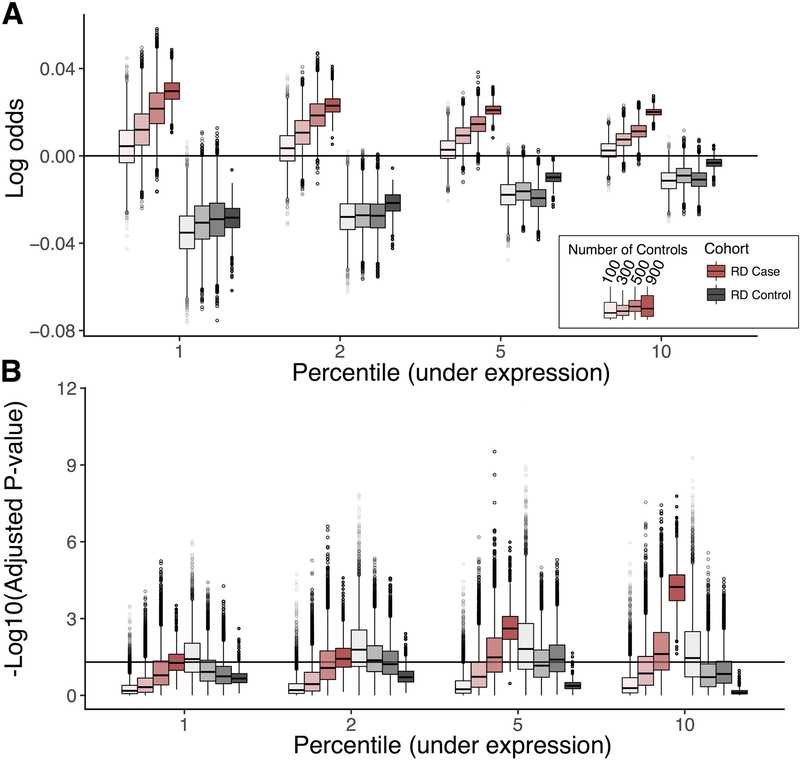
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
