

An update on the CNS manifestations of neurofibromatosis type 2
- PMID: 31161239
- PMCID: PMC7038792
- DOI: 10.1007/s00401-019-02029-5
An update on the CNS manifestations of neurofibromatosis type 2
Erratum in
-
Correction to: An update on the CNS manifestations of neurofibromatosis type 2.Acta Neuropathol. 2020 Apr;139(4):667. doi: 10.1007/s00401-019-02044-6. Acta Neuropathol. 2020. PMID: 31432207 Free PMC article.
Abstract
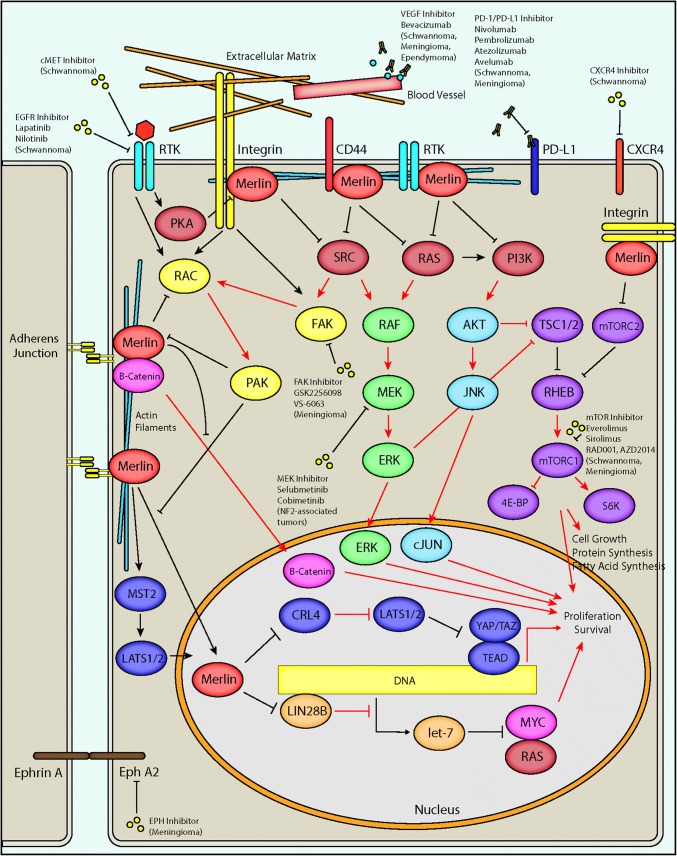
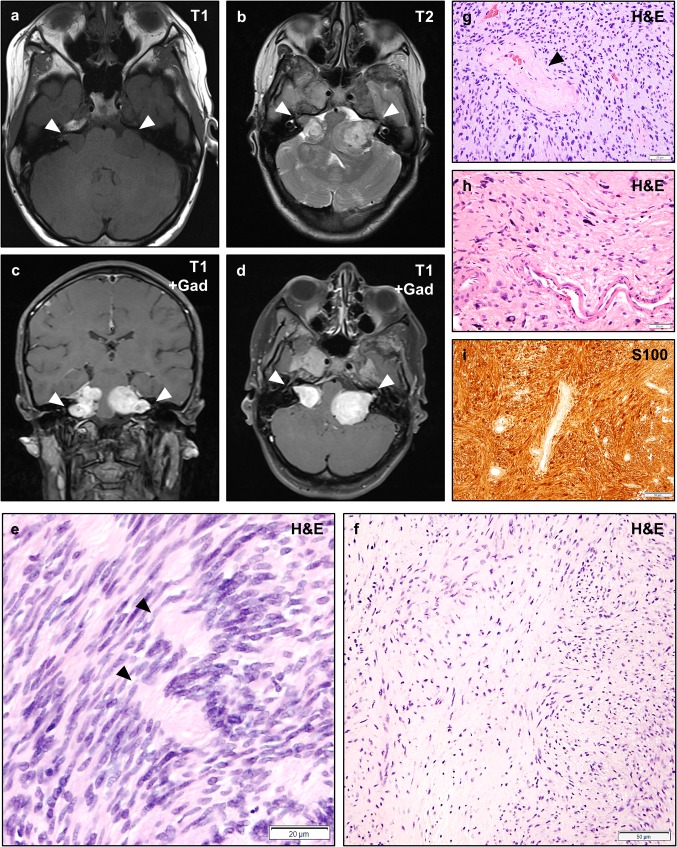
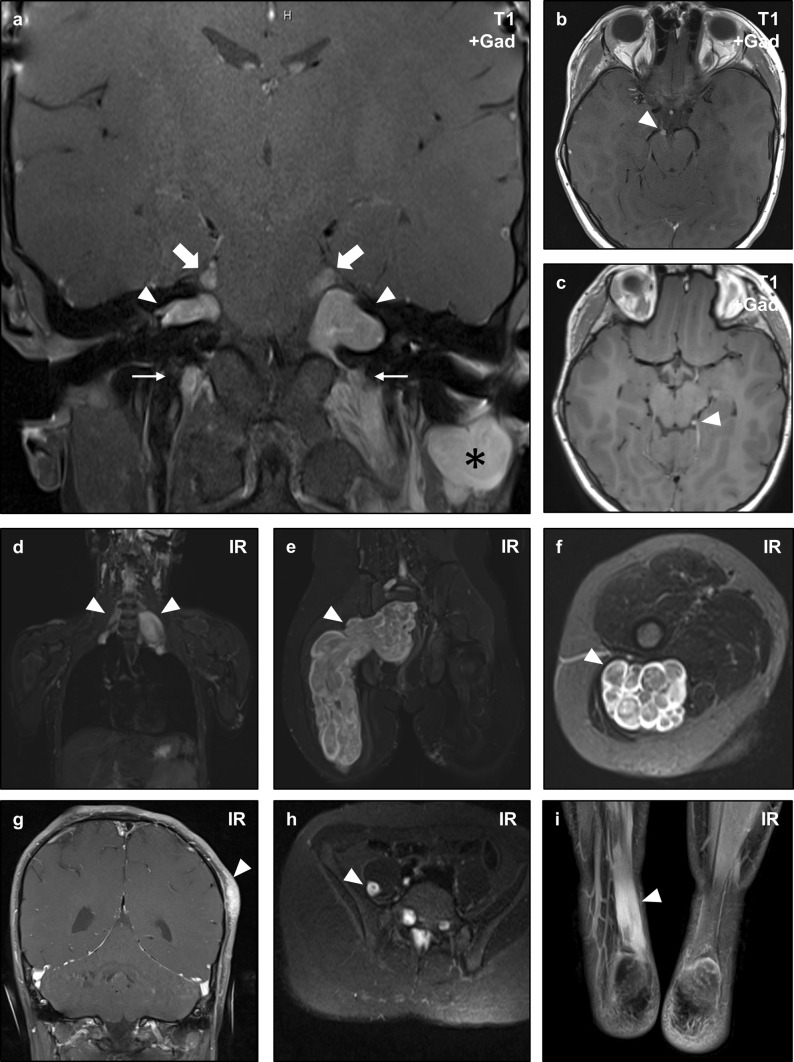
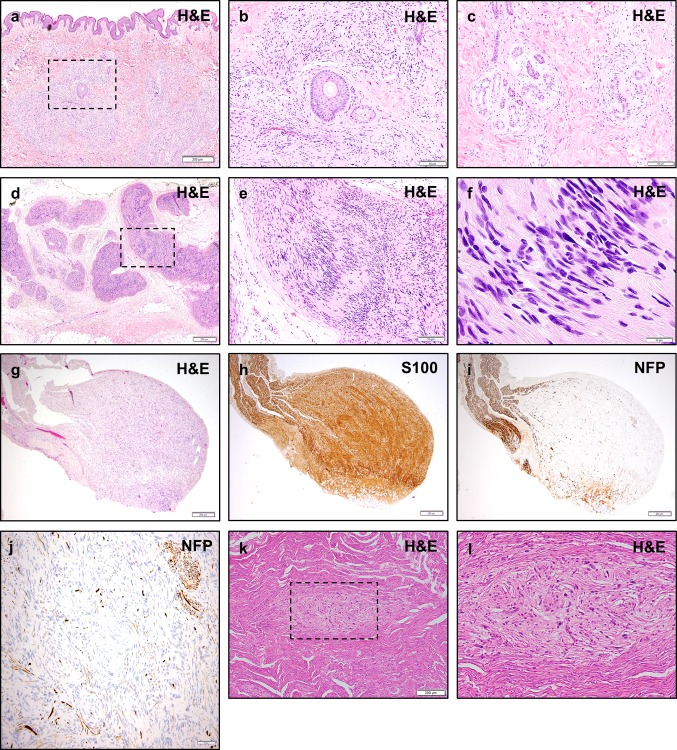
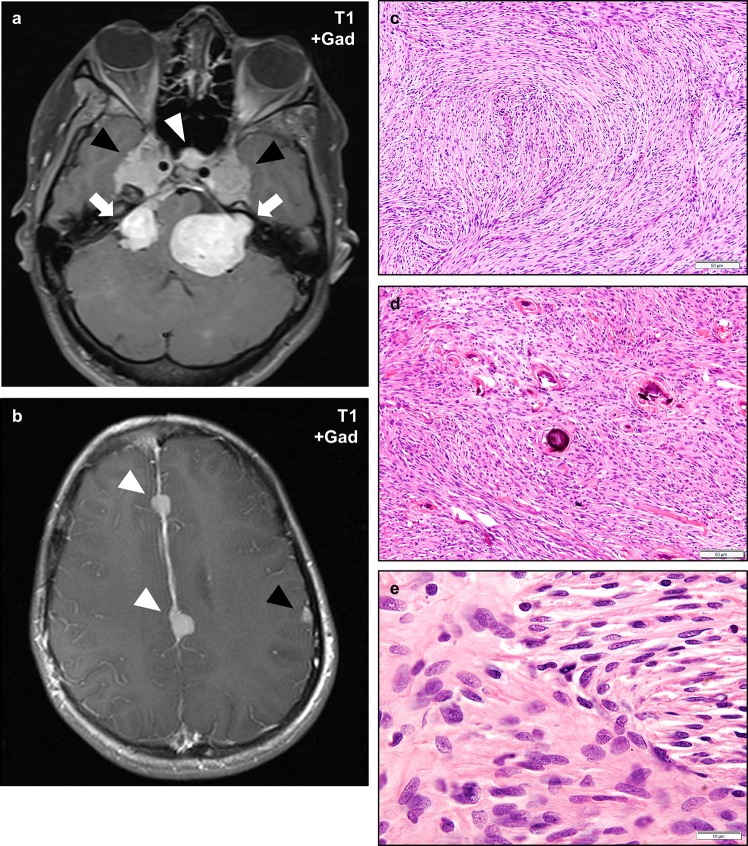
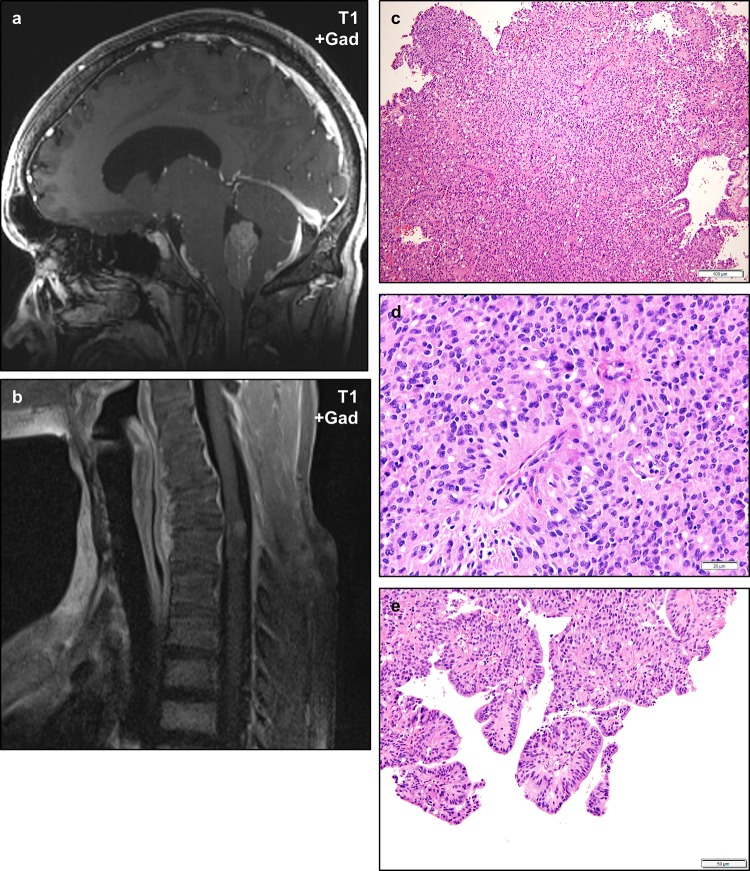
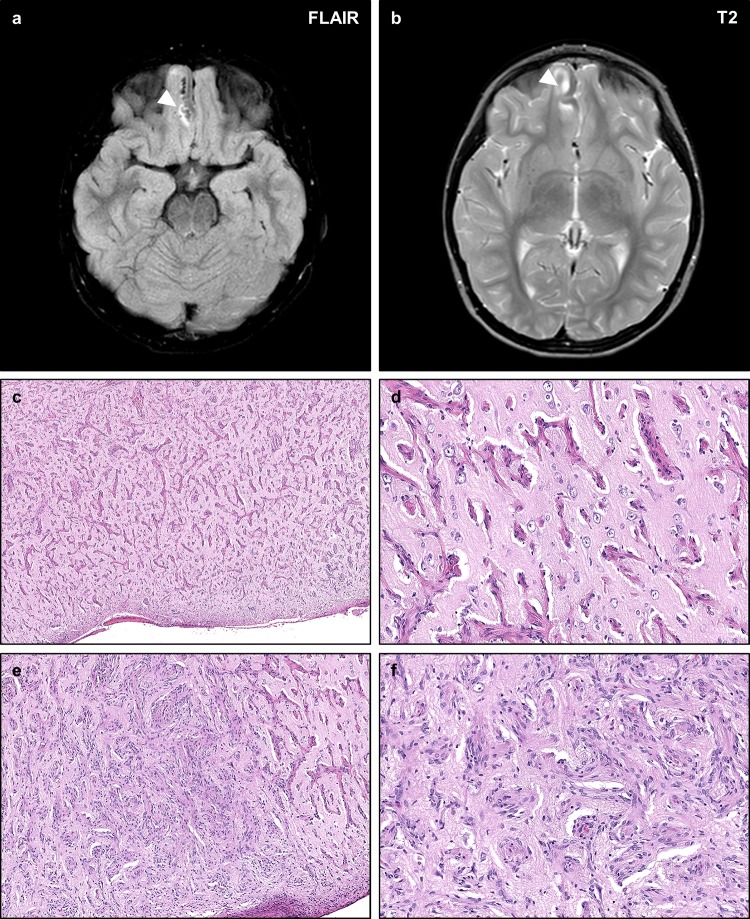
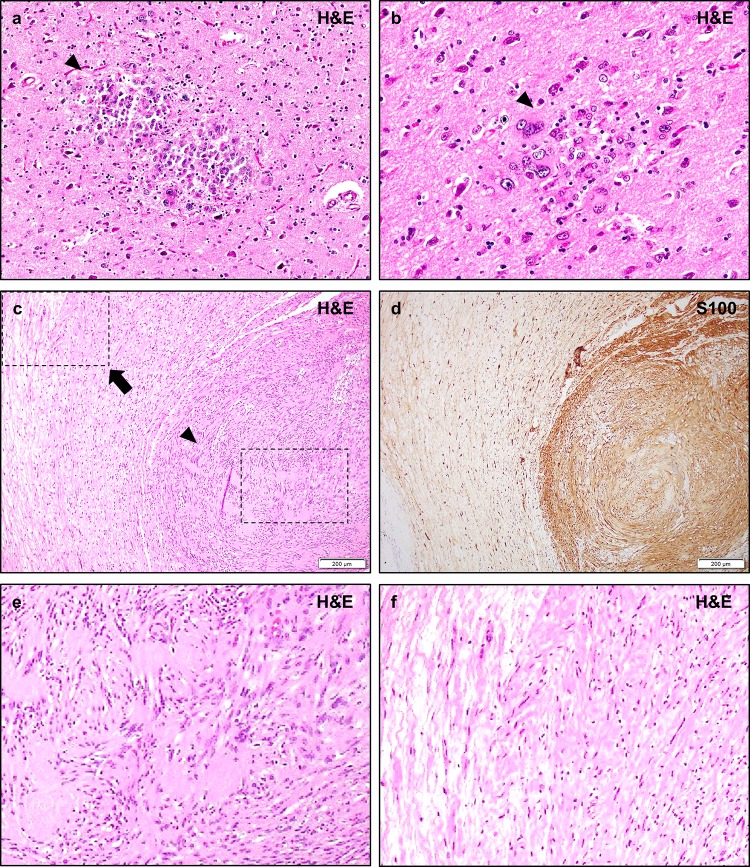
Neurofibromatosis type II (NF2) is a tumor predisposition syndrome characterized by the development of distinctive nervous system lesions. NF2 results from loss-of-function alterations in the NF2 gene on chromosome 22, with resultant dysfunction of its protein product merlin. NF2 is most commonly associated with the development of bilateral vestibular schwannomas; however, patients also have a predisposition to development of other tumors including meningiomas, ependymomas, and peripheral, spinal, and cranial nerve schwannomas. Patients may also develop other characteristic manifestations such as ocular lesions, neuropathies, meningioangiomatosis, and glial hamartia. NF2 has a highly variable clinical course, with some patients exhibiting a severe phenotype and development of multiple tumors at an early age, while others may be nearly asymptomatic throughout their lifetime. Despite the high morbidity associated with NF2 in severe cases, management of NF2-associated lesions primarily consists of surgical resection and treatment of symptoms, and there are currently no FDA-approved systemic therapies that address the underlying biology of the syndrome. Refinements to the diagnostic criteria of NF2 have been proposed over time due to increasing understanding of clinical and molecular data. Large-population studies have demonstrated that some features such as the development of gliomas and neurofibromas, currently included as diagnostic criteria, may require further clarification and modification. Meanwhile, burgeoning insights into the molecular biology of NF2 have shed light on the etiology and highly variable severity of the disease and suggested numerous putative molecular targets for therapeutic intervention. Here, we review the clinicopathologic features of NF2, current understanding of the molecular biology of NF2, particularly with regard to central nervous system lesions, ongoing therapeutic studies, and avenues for further research.
Keywords: Acoustic neuroma; Cellular schwannoma; Central neurofibromatosis; ERM (ezrin/radixin/moesin) family scaffolding; Ependymoma; Epidemiology of familial tumor syndromes; Gardner; Glial hamartia; Glial micro-hamartoma; Glioma; Intraneural schwannoma; LZTR1; Manchester (NIH) Criteria; Manchester Criteria; Meningioangiomatosis; Meningioma; Merlin; NF2; Neurofibroma; Neurofibromatosis type 2; Neurofibromatosis type II; Neurofibromin 2; Plexiform schwannoma; Posterior subcapsular lenticular opacities; SH3PXD2A-HTRA1; SMARCB1; SMARCE1; SUFU; Schwannoma; Schwannomatosis; Schwannomin; Verocay body; Vestibular schwannoma; Von Recklinghausen; Wishart.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO classification of tumors of the central nervous system. Lyon: IARC/WHO; 2016. pp. 294–303. - PubMed
-
- Riccardi VM. Neurofibromatosis: clinical heterogeneity. Curr Probl Cancer. 1982;8:1–34. - PubMed
-
- Ruggieri M, Praticò AD, Caltabiano R, Polizzi A. Early history of the different forms of neurofibromatosis from ancient Egypt to the British Empire and beyond: first descriptions, medical curiosities, misconceptions, landmarks, and the persons behind the syndromes. Am J Med Genet A. 2018;176(3):515–550. doi: 10.1002/ajmg.a.38486. - DOI - PubMed
-
- Von Recklinghausen F. Ueber die multiplen fibroma der haut und ihre beziehung zu den multiplen neuromen. Berlin: A Hirschwald; 1882. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
