

TAR-VIR: a pipeline for TARgeted VIRal strain reconstruction from metagenomic data
- PMID: 31164077
- PMCID: PMC6549370
- DOI: 10.1186/s12859-019-2878-2
TAR-VIR: a pipeline for TARgeted VIRal strain reconstruction from metagenomic data
Abstract
Background: Strain-level RNA virus characterization is essential for developing prevention and treatment strategies. Viral metagenomic data, which can contain sequences of both known and novel viruses, provide new opportunities for characterizing RNA viruses. Although there are a number of pipelines for analyzing viruses in metagenomic data, they have different limitations. First, viruses that lack closely related reference genomes cannot be detected with high sensitivity. Second, strain-level analysis is usually missing.
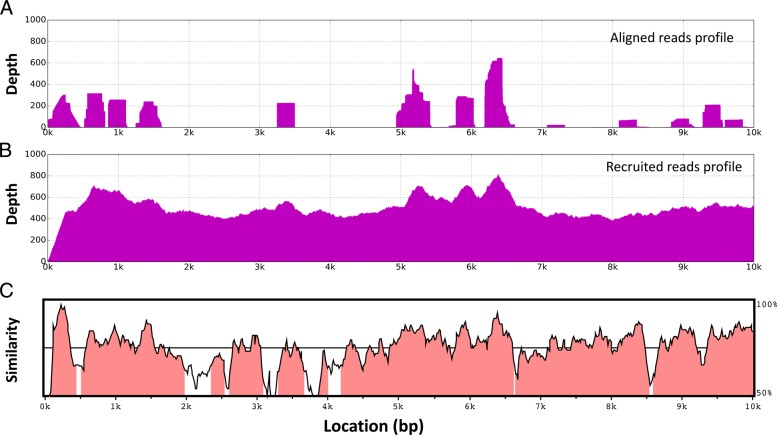
Results: In this study, we developed a hybrid pipeline named TAR-VIR that reconstructs viral strains without relying on complete or high-quality reference genomes. It is optimized for identifying RNA viruses from metagenomic data by combining an effective read classification method and our in-house strain-level de novo assembly tool. TAR-VIR was tested on both simulated and real viral metagenomic data sets. The results demonstrated that TAR-VIR competes favorably with other tested tools.
Conclusion: TAR-VIR can be used standalone for viral strain reconstruction from metagenomic data. Or, its read recruiting stage can be used with other de novo assembly tools for superior viral functional and taxonomic analyses. The source code and the documentation of TAR-VIR are available at https://github.com/chjiao/TAR-VIR .
Keywords: RNA virus; Read classification; Strain assembly; Viral metagenomics.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
