

Epithelial to mesenchymal transition (EMT) is associated with attenuation of succinate dehydrogenase (SDH) in breast cancer through reduced expression of SDHC
- PMID: 31164982
- PMCID: PMC6544948
- DOI: 10.1186/s40170-019-0197-8
Epithelial to mesenchymal transition (EMT) is associated with attenuation of succinate dehydrogenase (SDH) in breast cancer through reduced expression of SDHC
Abstract
Background: Epithelial to mesenchymal transition (EMT) is a well-characterized process of cell plasticity that may involve metabolic rewiring. In cancer, EMT is associated with malignant progression, tumor heterogeneity, and therapy resistance. In this study, we investigated the role of succinate dehydrogenase (SDH) as a potential key regulator of EMT.
Methods: Associations between SDH subunits and EMT were explored in gene expression data from breast cancer patient cohorts, followed by in-depth studies of SDH suppression as a potential mediator of EMT in cultured cells.
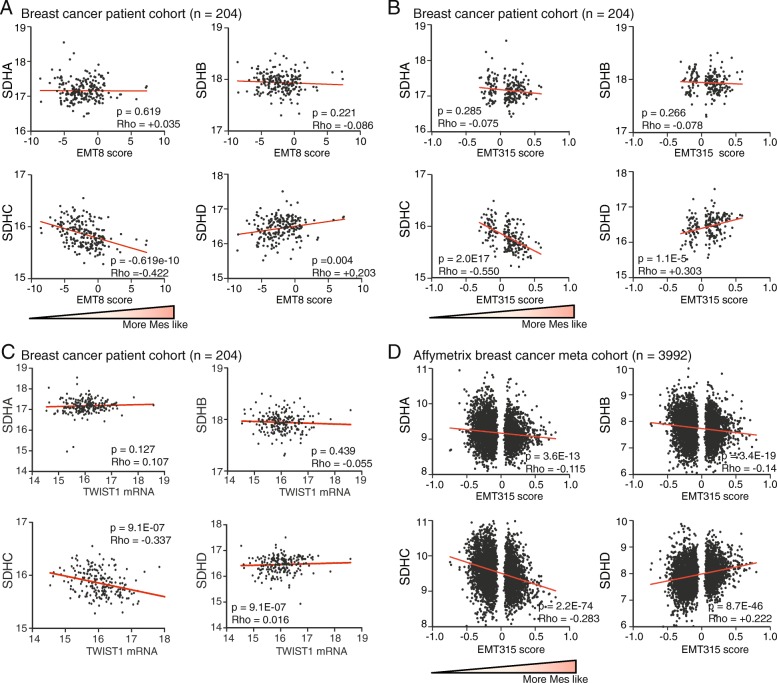
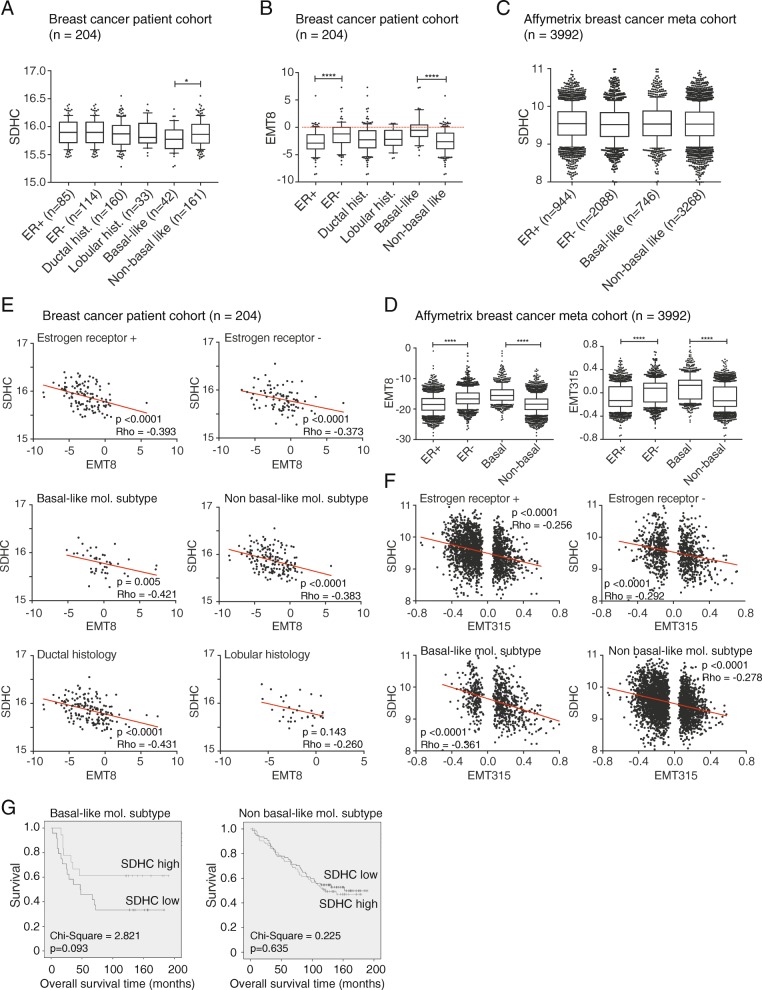
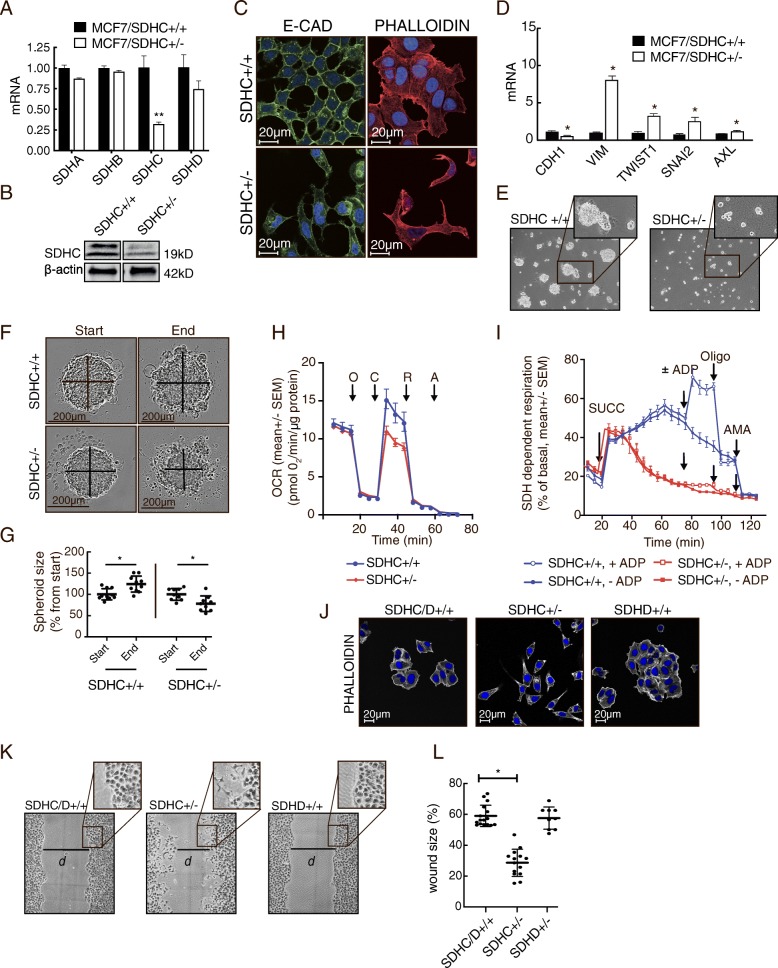
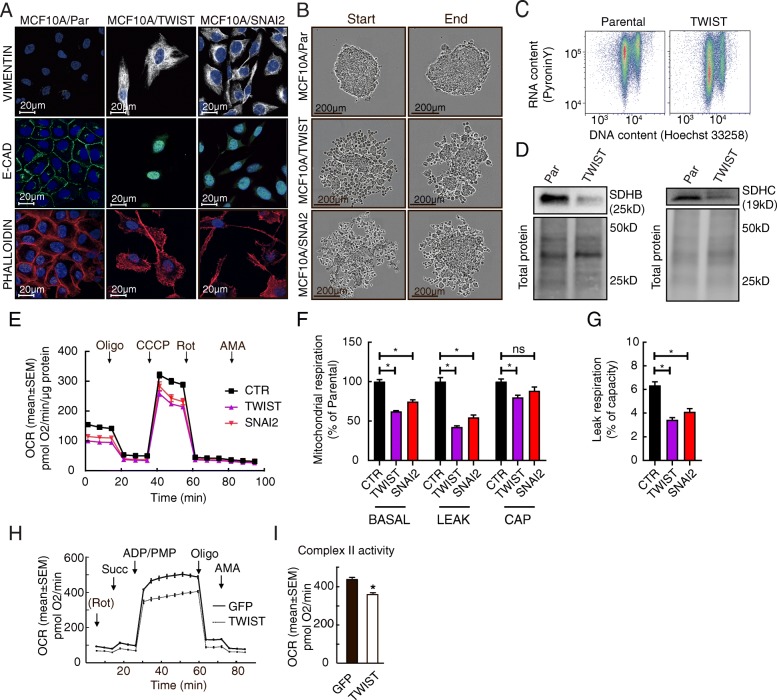
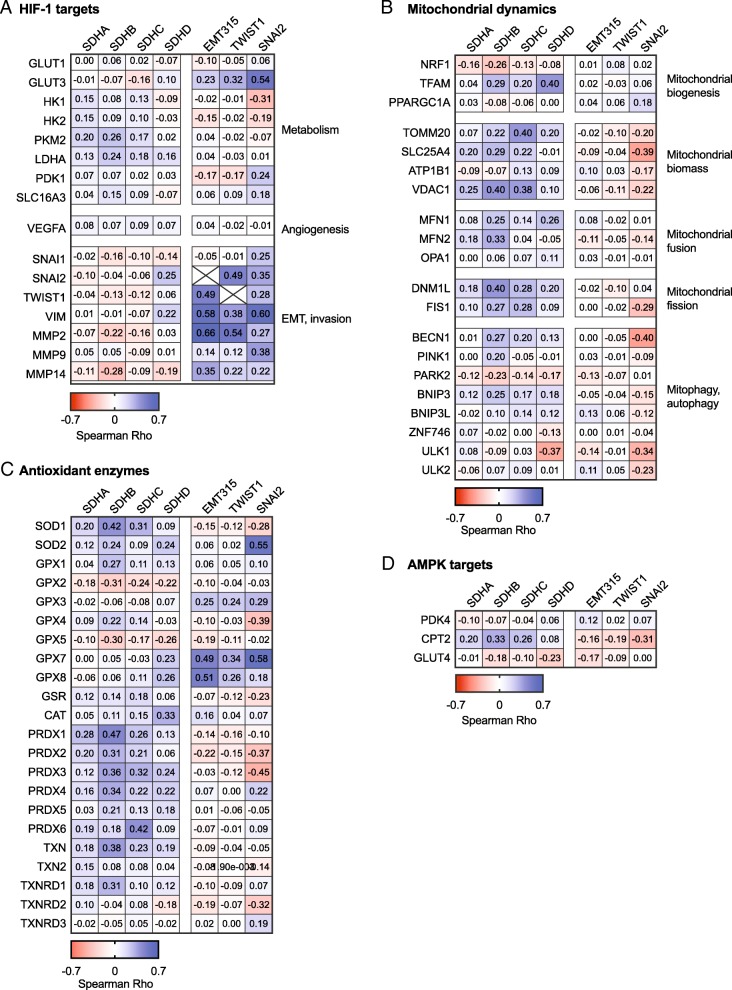
Results: We found an overall inverse association between EMT and the SDH subunit C (SDHC) when analyzing gene expression in breast tumors. This was particularly evident in carcinomas of basal-like molecular subtype compared to non-basal-like tumors, and a low SDHC expression level tended to have a prognostic impact in those patients. Studies in cultured cells revealed that EMT was induced by SDH inhibition through SDHC CRISPR/Cas9 knockdown or by the enzymatic inhibitor malonate. Conversely, overexpression of EMT-promoting transcription factors TWIST and SNAI2 caused decreased levels of SDHB and C and reduced rates of SDH-linked mitochondrial respiration. Cells overexpressing TWIST had reduced mitochondrial mass, and the organelles were thinner and more fragmented compared to controls.
Conclusions: Our findings suggest that downregulation of SDHC promotes EMT and that this is accompanied by structural remodeling of the mitochondrial organelles. This may confer survival benefits upon exposure to hostile microenvironment including oxidative stress and hypoxia during cancer progression.
Keywords: Breast cancer; Cell metabolism; Cell plasticity; Mitochondria; SDH.
Conflict of interest statement
Competing interestsThe authors declare that they have no competing interests.
Figures


References
LinkOut - more resources
Full Text Sources
Research Materials
