

Steric Switching From Photochemical to Thermal N2 Splitting: A Computational Analysis of the Isomerization Reaction {(Cp*)(Am)Mo}2(μ-η1:η1-N2) → {(Cp*)(Am)Mo}2(μ-N)2
- PMID: 31165063
- PMCID: PMC6535493
- DOI: 10.3389/fchem.2019.00352
Steric Switching From Photochemical to Thermal N2 Splitting: A Computational Analysis of the Isomerization Reaction {(Cp*)(Am)Mo}2(μ-η1:η1-N2) → {(Cp*)(Am)Mo}2(μ-N)2
Abstract
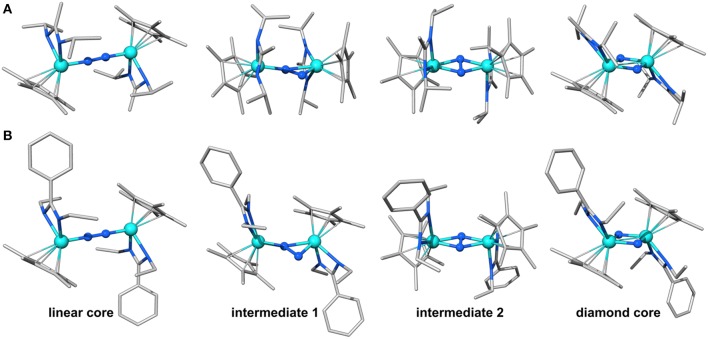
A μ-η1:η1-N2-bridged Mo dimer, {(η5-C5Me5)[N(Et)C(Ph)N(Et)]Mo}2(μ-N2), cleaves dinitrogen thermally resulting in a crystallographically characterized bis-μ-N-bridged dimer, {(η5-C5Me5)[N(Et)C(Ph)N(Et)]Mo}2(μ-N)2. A structurally related Mo dimer with a bulkier amidinate ligand, ([N(iPr)C(Me)N(iPr)]), is only capable of photochemical dinitrogen activation. These opposing reactivities were rationalized as steric switching between the thermally and photochemically active species. A computational analysis of the geometric and electronic structures of intermediates along the isomerization pathway from Mo2(μ-η1:η1-N2) to Mo2(μ-η2:η1-N2) and Mo2(μ-η2:η2-N2), and finally Mo2(μ-N)2, is presented here. The extent to which dispersion affects the thermodynamics of the isomers is evaluated, and it is found that dispersion interactions play a significant role in stabilizing the product and making the reaction exergonic. The concept of steric switching is further explored with theoretical models with sterically even less demanding ligands, indicating that systematic ligand modifications could be used to rationally design the N2 activation energy landscape. An analysis of electronic excitations in the computed UV-vis spectra of the two complexes shows that a particular type of asymmetric excitations is only present in the photoactive complex.
Keywords: density functional theory; isomerization thermodynamics; molybdenum; nitrogen fixation; theoretical UV-vis spectroscopy.
Figures








References
-
- Adamo C., Barone V. (1999). Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170. 10.1063/1.478522 - DOI
-
- Andrae D., Haussermann U., Dolg M., Stoll H., Preuss H. (1990). Energy-adjusted Ab initio pseudopotentials for the 2nd and 3rd row transition-elements. Theor. Chim. Acta 77, 123–141.
-
- Becke A. D. (1988). Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 38, 3098–3100. - PubMed
-
- Becke A. D. (1993). Density-functional thermochemistry.3. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. 10.1063/1.464913 - DOI
-
- Broda H., Hinrichsen S., Tuczek F. (2013). Molybdenum(0) dinitrogen complexes with polydentate phosphine ligands for synthetic nitrogen fixation: geometric and electronic structure contributions to reactivity. Coord. Chem. Rev. 257, 587–598. 10.1016/j.ccr.2012.05.010 - DOI
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous