

Phosphorylation of FANCD2 Inhibits the FANCD2/FANCI Complex and Suppresses the Fanconi Anemia Pathway in the Absence of DNA Damage
- PMID: 31167143
- PMCID: PMC6581795
- DOI: 10.1016/j.celrep.2019.05.003
Phosphorylation of FANCD2 Inhibits the FANCD2/FANCI Complex and Suppresses the Fanconi Anemia Pathway in the Absence of DNA Damage
Abstract
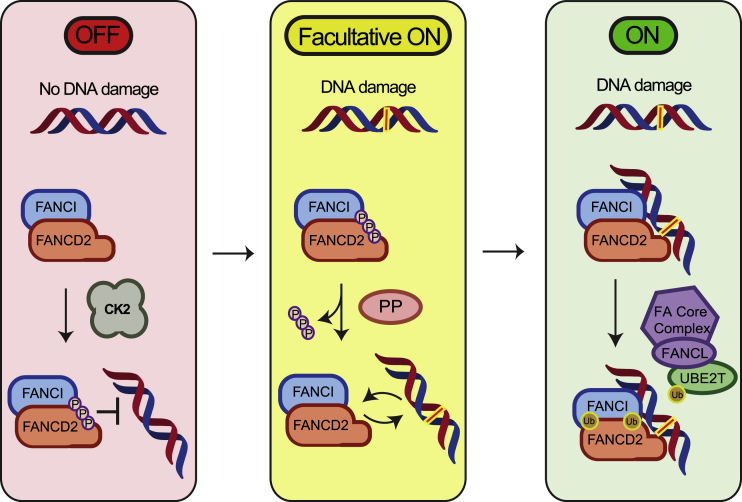
Interstrand crosslinks (ICLs) of the DNA helix are a deleterious form of DNA damage. ICLs can be repaired by the Fanconi anemia pathway. At the center of the pathway is the FANCD2/FANCI complex, recruitment of which to DNA is a critical step for repair. After recruitment, monoubiquitination of both FANCD2 and FANCI leads to their retention on chromatin, ensuring subsequent repair. However, regulation of recruitment is poorly understood. Here, we report a cluster of phosphosites on FANCD2 whose phosphorylation by CK2 inhibits both FANCD2 recruitment to ICLs and its monoubiquitination in vitro and in vivo. We have found that phosphorylated FANCD2 possesses reduced DNA binding activity, explaining the previous observations. Thus, we describe a regulatory mechanism operating as a molecular switch, where in the absence of DNA damage, the FANCD2/FANCI complex is prevented from loading onto DNA, effectively suppressing the FA pathway.
Keywords: CK2; DNA repair; FANCD2/FANCI; Fanconi anemia; ICL repair; casein kinase 2; genome stability; interstrand crosslink repair; kinase; phosphorylation.
Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures






References
-
- Andreassen P.R., D’Andrea A.D., Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18:1958–1963. - PMC - PubMed
- Andreassen, P.R., D’Andrea, A.D., and Taniguchi, T. (2004). ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 18, 1958-1963. - PMC - PubMed
-
- Arter M., Hurtado-Nieves V., Oke A., Zhuge T., Wettstein R., Fung J.C., Blanco M.G., Matos J. Regulated crossing-over requires inactivation of Yen1/GEN1 resolvase during meiotic prophase I. Dev. Cell. 2018;45:785–800.e6. - PMC - PubMed
- Arter, M., Hurtado-Nieves, V., Oke, A., Zhuge, T., Wettstein, R., Fung, J.C., Blanco, M.G., and Matos, J. (2018). Regulated crossing-over requires inactivation of Yen1/GEN1 resolvase during meiotic prophase I. Dev. Cell 45, 785-800.e6. - PMC - PubMed
-
- Ayoub N., Jeyasekharan A.D., Bernal J.A., Venkitaraman A.R. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–686. - PubMed
- Ayoub, N., Jeyasekharan, A.D., Bernal, J.A., and Venkitaraman, A.R. (2008). HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature 453, 682-686. - PubMed
-
- Blanco M.G., Matos J., West S.C. Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol. Cell. 2014;54:94–106. - PMC - PubMed
- Blanco, M.G., Matos, J., and West, S.C. (2014). Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol. Cell 54, 94-106. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
