

Complement Membrane Attack Complexes Assemble NLRP3 Inflammasomes Triggering IL-1 Activation of IFN-γ-Primed Human Endothelium
- PMID: 31170059
- PMCID: PMC6557295
- DOI: 10.1161/CIRCRESAHA.119.314845
Complement Membrane Attack Complexes Assemble NLRP3 Inflammasomes Triggering IL-1 Activation of IFN-γ-Primed Human Endothelium
Erratum in
-
Correction to: Complement Membrane Attack Complexes Assemble NLRP3 Inflammasomes Triggering IL-1 Activation of IFN-γ-Primed Human Endothelium.Circ Res. 2021 Apr 16;128(8):e122. doi: 10.1161/RES.0000000000000472. Epub 2021 Apr 15. Circ Res. 2021. PMID: 33856920 No abstract available.
Abstract
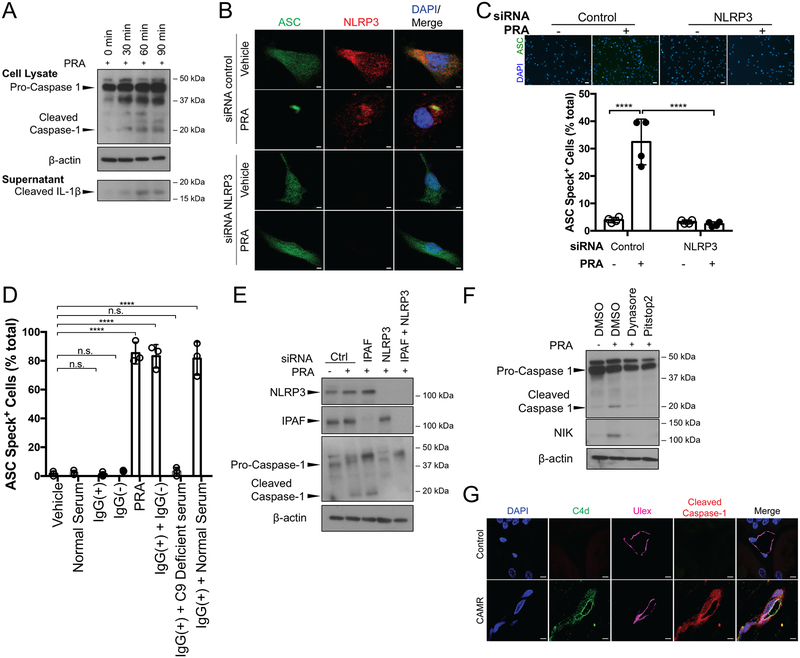
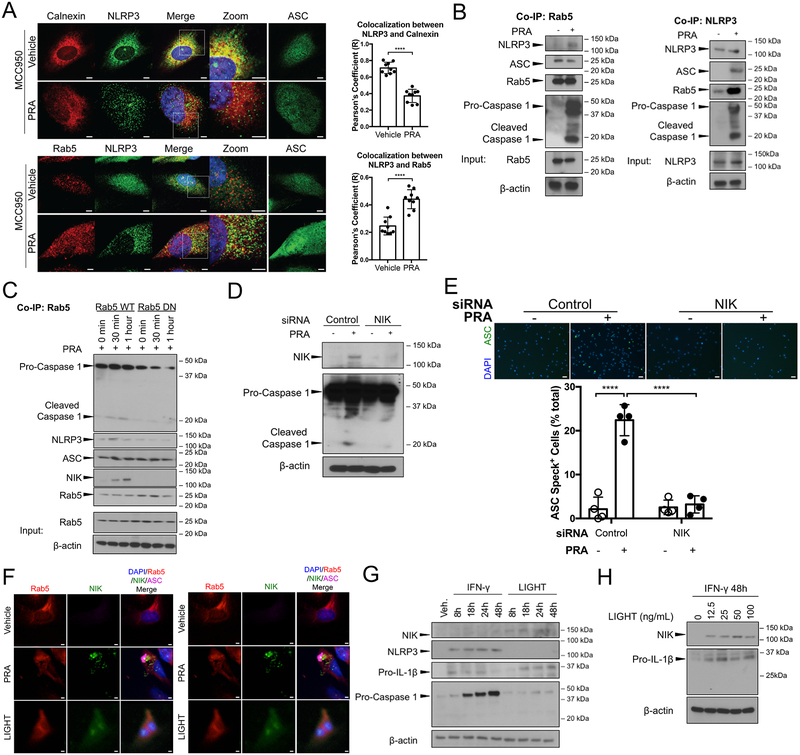
Rationale: Complement activation contributes to multiple immune-mediated pathologies. In late allograft failure, donor-specific antibody deposits complement membrane attack complexes (MAC) on graft endothelial cells (ECs), substantially increasing their immunogenicity without causing lysis. Internalized MAC stabilize NIK (NF-κB [nuclear factor kappa-light-chain-enhancer of activated B cells]-inducing kinase) protein on Rab5+MAC+ endosomes, activating noncanonical NF-κB signaling. However, the link to increased immunogenicity is unclear.
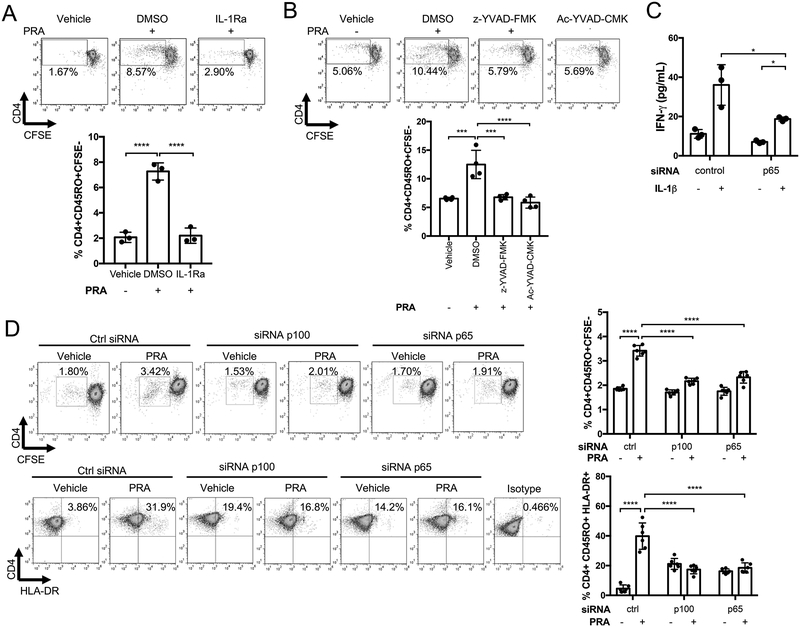
Objective: To identify mechanisms by which alloantibody and internalized MAC activate ECs to enhance their ability to increase T-cell responses.
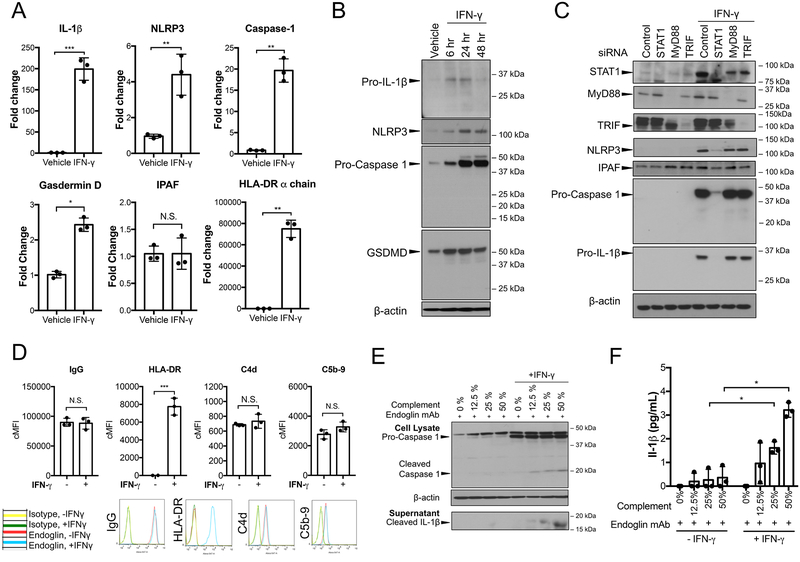
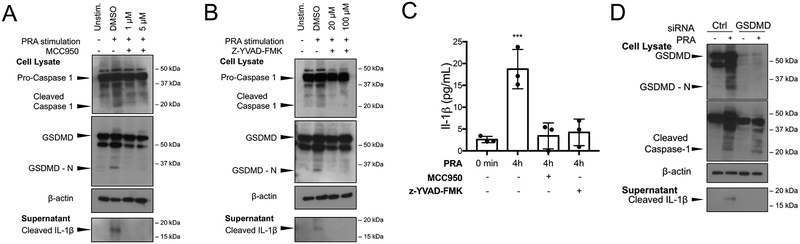
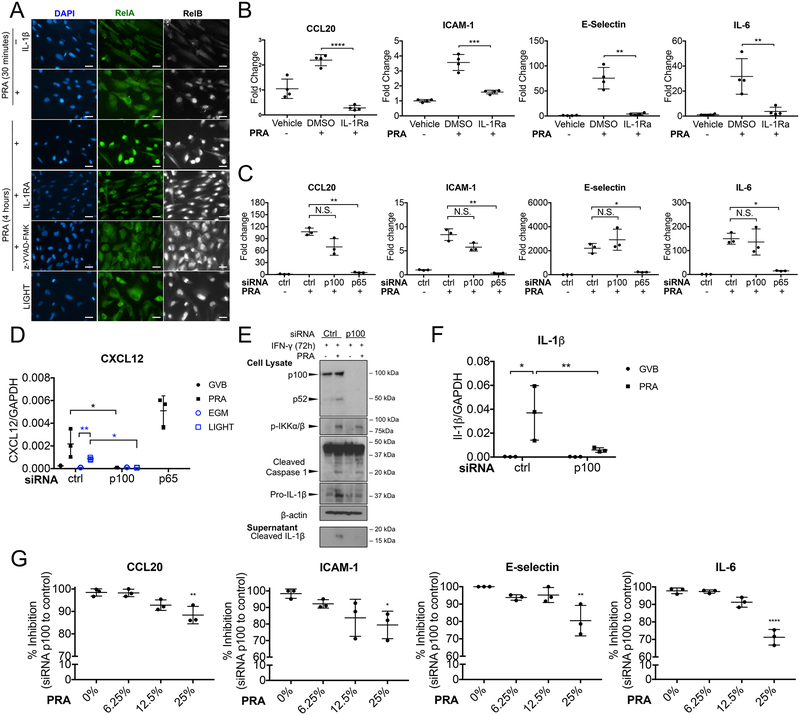
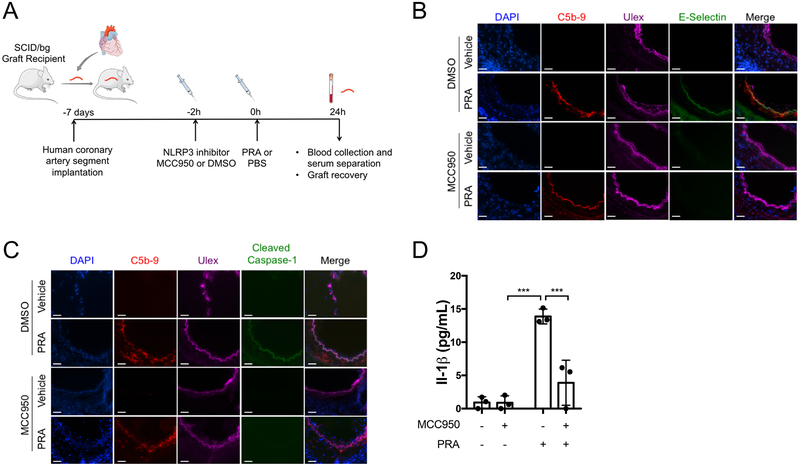
Methods and results: In human EC cultures, internalized MAC also causes NLRP3 (NOD-like receptor family pyrin domain containing 3) translocation from endoplasmic reticulum to Rab5+MAC+NIK+ endosomes followed by endosomal NIK-dependent inflammasome assembly. Cytosolic NIK, stabilized by LIGHT (lymphotoxin-like inducible protein that competes with glycoprotein D for herpesvirus entry on T cells), does not trigger inflammasome assembly, and ATP-triggered inflammasome assembly does not require NIK. IFN-γ (interferon-γ) primes EC responsiveness to MAC by increasing NLRP3, pro-caspase 1, and gasdermin D expression. NIK-activated noncanonical NF-κB signaling induces pro-IL (interleukin)-1β expression. Inflammasome processed pro-IL-1β, and gasdermin D results in IL-1β secretion that increases EC immunogenicity through IL-1 receptor signaling. Activation of human ECs lining human coronary artery grafts in immunodeficient mouse hosts by alloantibody and complement similarly depends on assembly of an NLRP3 inflammasome. Finally, in renal allograft biopsies showing chronic rejection, caspase-1 is activated in C4d+ ECs of interstitial microvessels, supporting the relevance of the cell culture findings.
Conclusions: In response to antibody-mediated complement activation, IFN-γ-primed human ECs internalize MAC, triggering both endosomal-associated NIK-dependent NLRP3 inflammasome assembly and IL-1 synthesis, resulting in autocrine/paracrine IL-1β-mediated increases in EC immunogenicity. Similar responses may underlie other complement-mediated pathologies.
Keywords: endosomes; endothelium; immune system; inflammasomes; transplantation.
Figures
References
-
- Abrahimi P, Liu R, Pober JS. Blood Vessels in Allotransplantation. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2015;15:1748–1754 - PubMed
-
- Jane-Wit D, Manes TD, Yi T, Qin L, Clark P, Kirkiles-Smith NC, Abrahimi P, Devalliere J, Moeckel G, Kulkarni S, Tellides G, Pober JS. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-kappaB signaling in endothelial cells. Circulation. 2013;128:2504–2516 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
