

PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses
- PMID: 31170232
- PMCID: PMC6553779
- DOI: 10.1371/journal.pone.0218003
PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses
Abstract
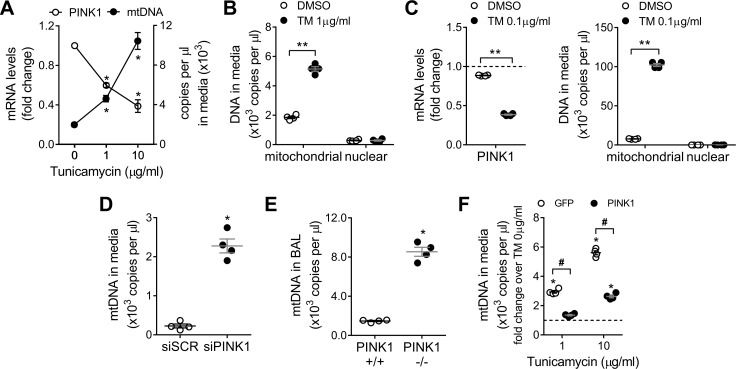
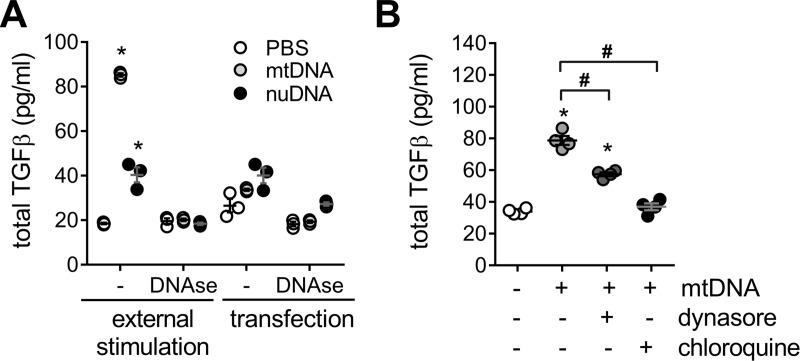
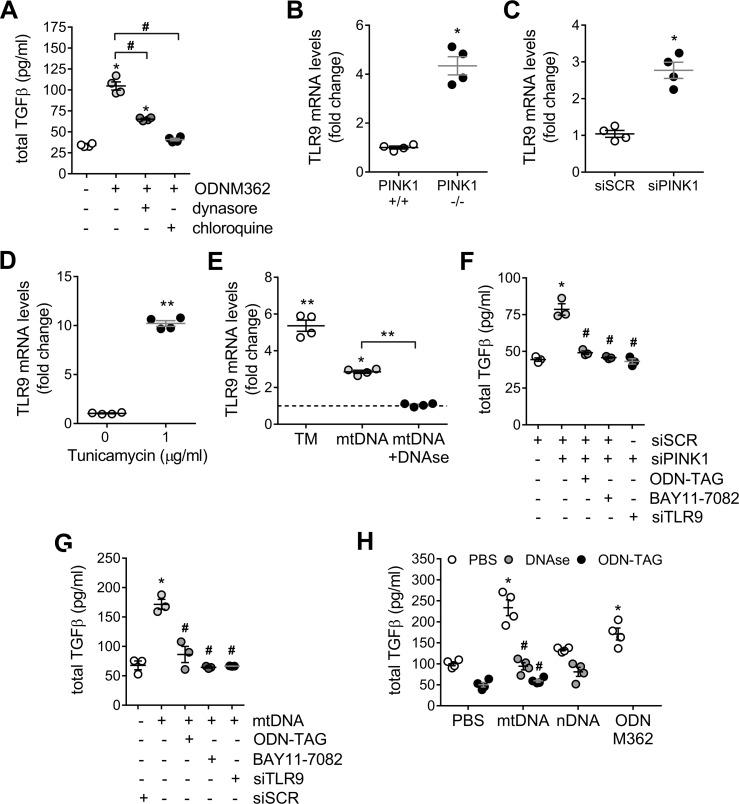
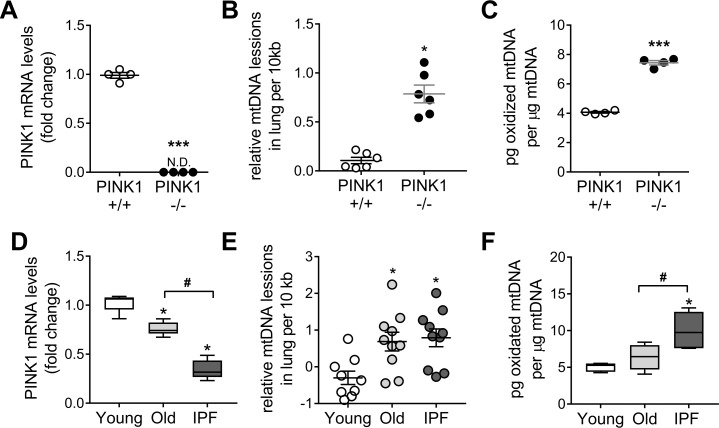
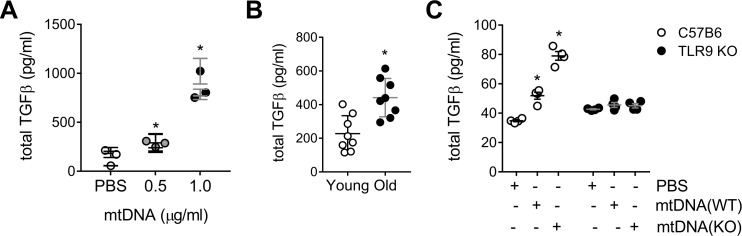
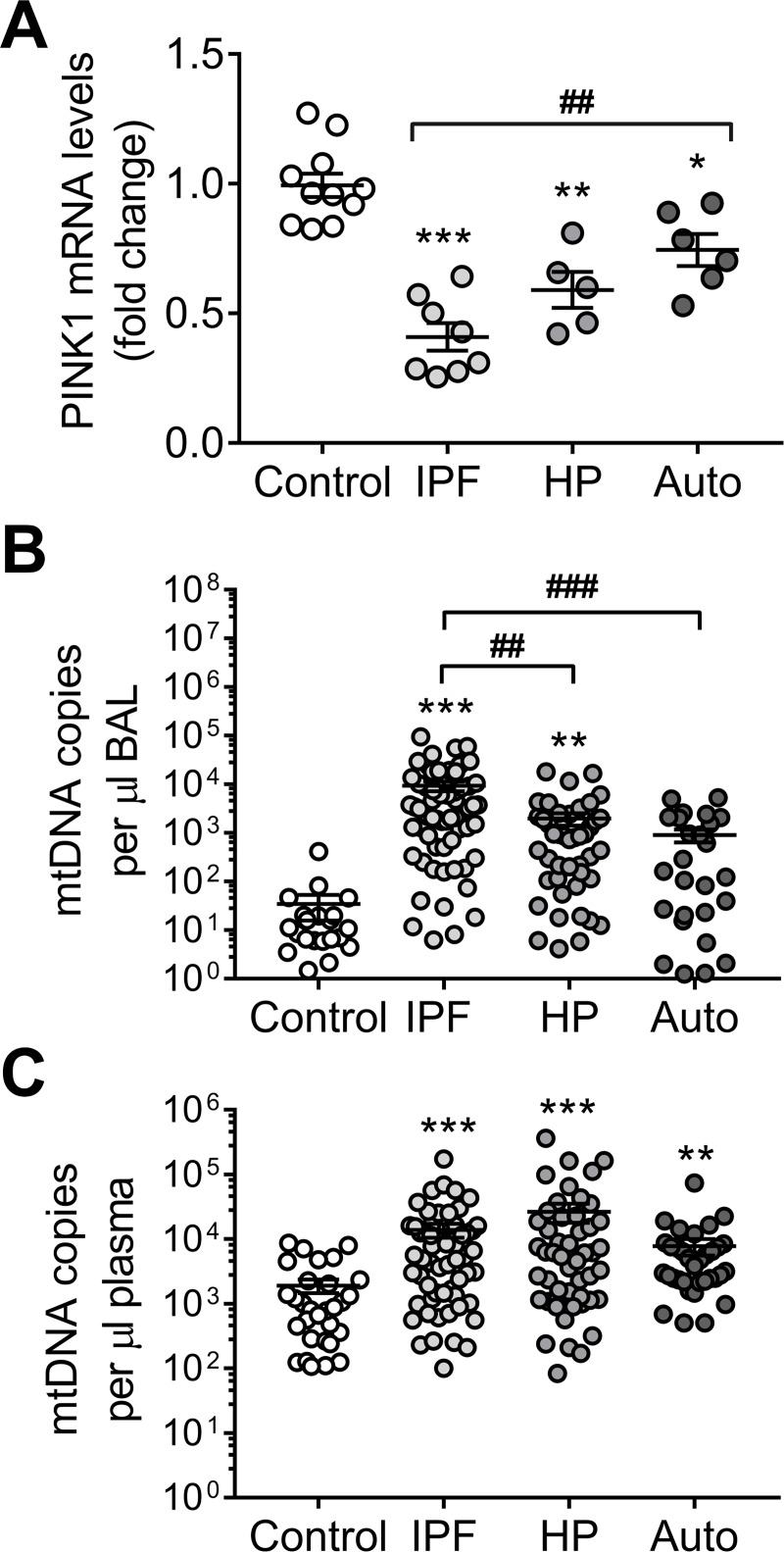
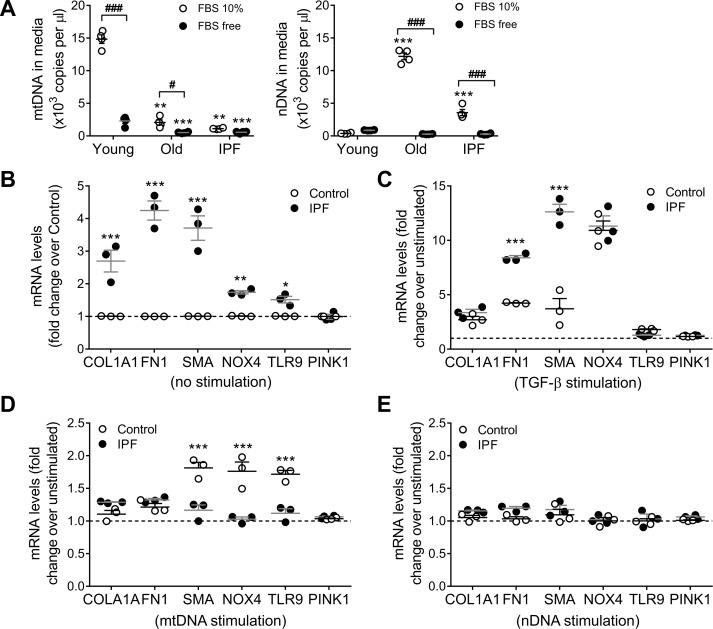
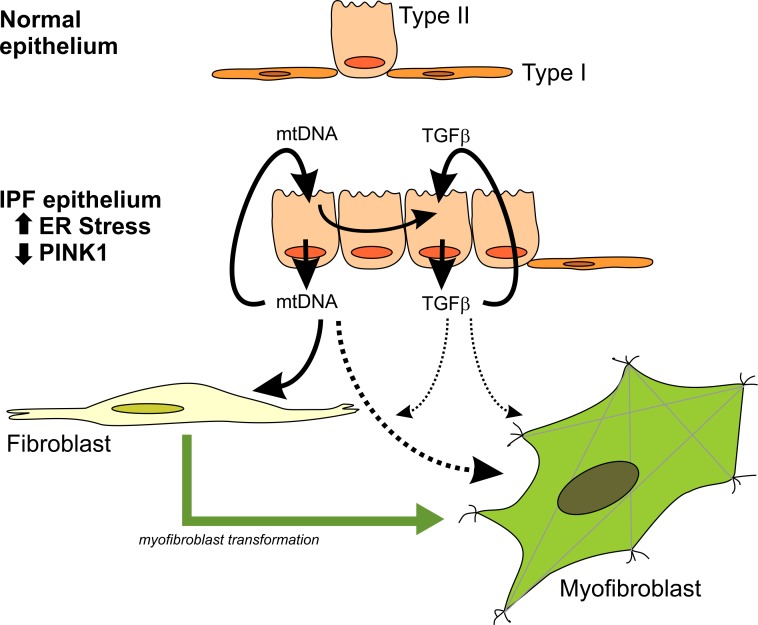
We have previously shown that endoplasmic reticulum stress (ER stress) represses the PTEN inducible kinase 1 (PINK1) in lung type II alveolar epithelial cells (AECII) reducing mitophagy and increasing the susceptibility to lung fibrosis. Although increased circulating mitochondrial DNA (mtDNA) has been reported in chronic lung diseases, the contribution of mitophagy in the modulation of mitochondrial DAMP release and activation of profibrotic responses is unknown. In this study, we show that ER stress and PINK1 deficiency in AECII led to mitochondrial stress with significant oxidation and damage of mtDNA and subsequent extracellular release. Extracellular mtDNA was recognized by TLR9 in AECII by an endocytic-dependent pathway. PINK1 deficiency-dependent mtDNA release promoted activation of TLR9 and triggered secretion of the profibrotic factor TGF-β which was rescued by PINK1 overexpression. Enhanced mtDNA oxidation and damage were found in aging and IPF human lungs and, in concordance, levels of circulating mtDNA were significantly elevated in plasma and bronchoalveolar lavage (BAL) from patients with IPF. Free mtDNA was found elevated in other ILDs with low expression of PINK1 including hypersensitivity pneumonitis and autoimmune interstitial lung diseases. These results support a role for PINK1 mediated mitophagy in the attenuation of mitochondrial damage associated molecular patterns (DAMP) release and control of TGF-β mediated profibrotic responses.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
