

α-Synuclein pathology in Parkinson's disease and related α-synucleinopathies
- PMID: 31170426
- PMCID: PMC7014913
- DOI: 10.1016/j.neulet.2019.134316
α-Synuclein pathology in Parkinson's disease and related α-synucleinopathies
Abstract
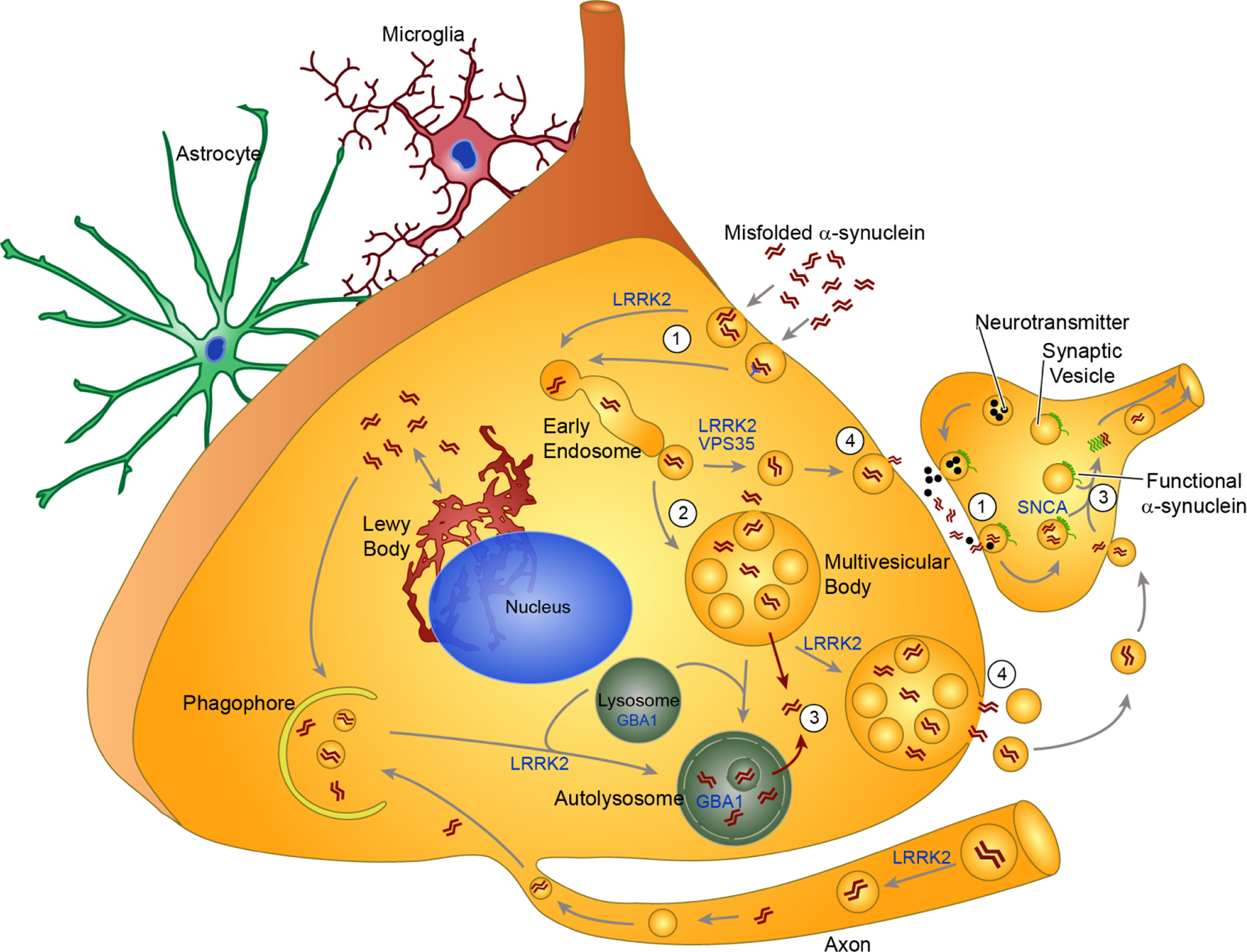
Over 20 years ago, the synaptic protein α-synuclein was identified as the primary component of the Lewy bodies (LBs) that are a sine qua non of Parkinson's disease (PD). Since that time, extensive research has demonstrated that α-synuclein pathology is not only a hallmark of PD, but can also cause neuronal dysfunction and death. Detailed staging of α-synuclein pathology in the brains of patients has revealed a progressive pattern of pathology that correlates with the symptoms of disease. Early in the disease course, PD patients exhibit motor dysfunction, and α-synuclein pathology at this stage is primarily found in regions controlling motor function. At later stages of disease as patients' cognitive function deteriorates, α-synuclein pathology can be found in cortical structures responsible for higher cognitive processing. The stereotypical progression of α-synuclein pathology through the brain over time suggests that there may be a physical transmission of pathological α-synuclein from one area of the brain to another. The transmission hypothesis posits that an initial seed of pathological α-synuclein in one neuron may be released and taken up by another vulnerable neuron and thereby initiate pathological misfolding of α-synuclein in the recipient neuron. In recent years, convergent evidence from various studies has indicated that pathological protein transmission can occur in the human brain. Cell and animal models based on the transmission hypothesis have shown not only that pathological α-synuclein can be transmitted from cell-to-cell, but that this pathology can lead to neuronal dysfunction and degeneration. The α-synuclein transmission hypothesis has profound implications for treatment of what is currently an intractable neurodegenerative disease. In this review, we explore the evidence for cell-to-cell transmission of pathological α-synuclein, the current understanding of how pathological α-synuclein can move to a new cell and template misfolding, and the therapeutic implications of α-synuclein transmission.
Keywords: Lewy body; Misfolding; Neurodegeneration; Prion-like; Transmission.
Copyright © 2019 Elsevier B.V. All rights reserved.
Figures


References
-
- Pringsheim T, et al., The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord, 2014. 29(13): p. 1583–90. - PubMed
-
- Parkinson J, An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci, 2002. 14(2): p. 223–36; discussion 222. - PubMed
-
- Poewe W, et al., Parkinson disease. Nat Rev Dis Primers, 2017. 3: p. 17013. - PubMed
-
- Dauer W and Przedborski S, Parkinson’s disease: mechanisms and models. Neuron, 2003. 39(6): p. 889–909. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
