

The genomic determinants of adaptive evolution in a fungal pathogen
- PMID: 31171985
- PMCID: PMC6546377
- DOI: 10.1002/evl3.117
The genomic determinants of adaptive evolution in a fungal pathogen
Abstract
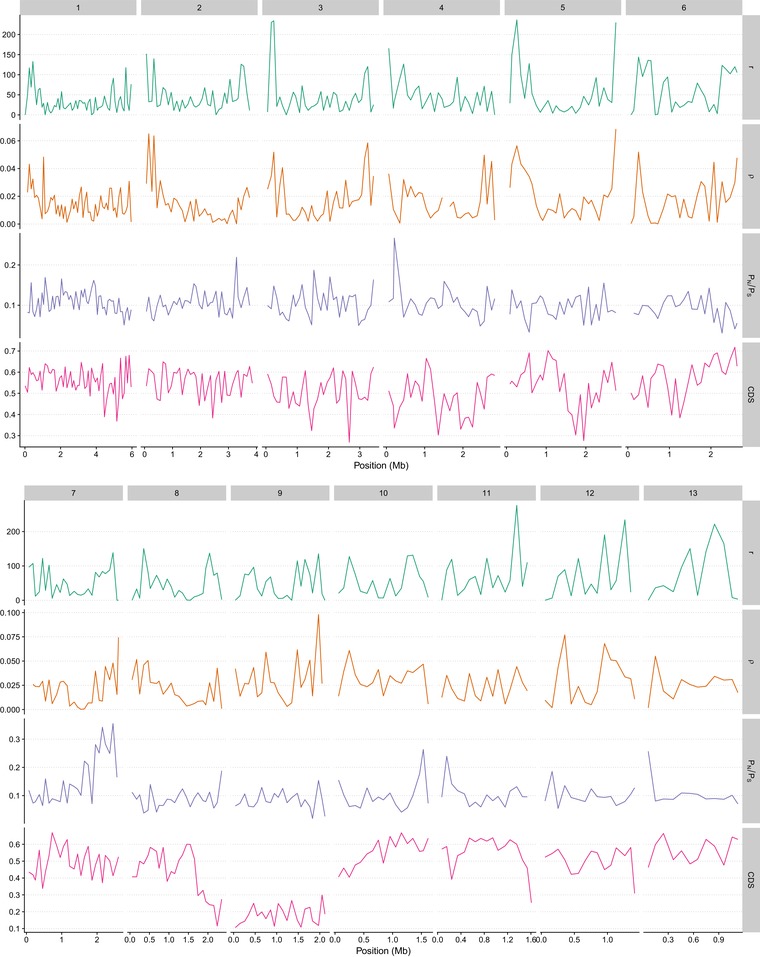
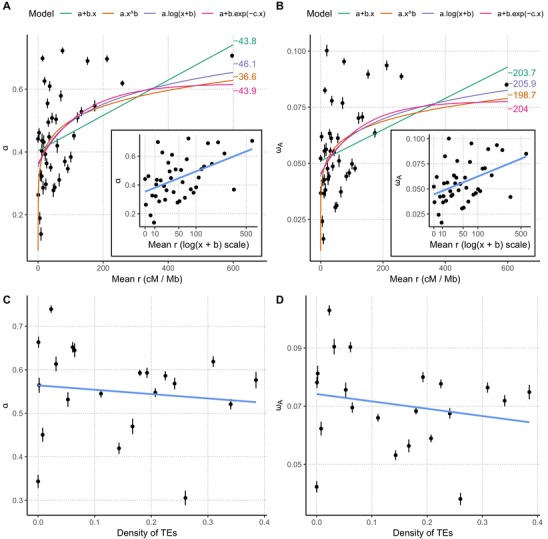
Unravelling the strength, frequency, and distribution of selective variants along the genome as well as the underlying factors shaping this distribution are fundamental goals of evolutionary biology. Antagonistic host-pathogen coevolution is thought to be a major driver of genome evolution between interacting species. While rapid evolution of pathogens has been documented in several model organisms, the genetic mechanisms of their adaptation are still poorly understood and debated, particularly the role of sexual reproduction. Here, we apply a population genomic approach to infer genome-wide patterns of selection among 13 isolates of Zymoseptoria tritici, a fungal pathogen characterized by extremely high genetic diversity, gene density, and recombination rates. We report that the genome of Z. tritici undergoes a high rate of adaptive substitutions, with 44% of nonsynonymous substitutions being adaptive on average. This fraction reaches 68% in so-called effector genes encoding determinants of pathogenicity, and the distribution of fitness effects differs in this class of genes as they undergo adaptive mutations with stronger positive fitness effects, but also more slightly deleterious mutations. Besides the globally high rate of adaptive substitutions, we report a negative relationship between pN/pS and the fine-scale recombination rate and a strong positive correlation between the rate of adaptive nonsynonymous substitutions (ωa) and recombination rate. This result suggests a pervasive role of both background selection and Hill-Robertson interference even in a species with an exceptionally high recombination rate (60 cM/Mb on average). While transposable elements (TEs) have been suggested to contribute to adaptation by creating compartments of fast-evolving genomic regions, we do not find a significant effect of TEs on the rate of adaptive mutations. Overall our study suggests that sexual recombination is a significant driver of genome evolution, even in rapidly evolving organisms subject to recurrent mutations with large positive effects.
Keywords: Adaptation; evolutionary rates; genome evolution; plant pathogenic fungi; recombination.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- Aguileta, G. , Lengelle J., Marthey S., Chiapello H., Rodolphe F., Gendrault A., et al. 2010. Finding candidate genes under positive selection in non‐model species: examples of genes involved in host specialization in pathogens. Mol. Ecol. 19:292–306. - PubMed
-
- Birdsell, J. A. 2002. Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol. Biol. Evol. 19:1181–1197. - PubMed
Associated data
LinkOut - more resources
Full Text Sources
Miscellaneous
