

Replication-Coupled DNA Repair
- PMID: 31173722
- PMCID: PMC6557297
- DOI: 10.1016/j.molcel.2019.04.027
Replication-Coupled DNA Repair
Abstract
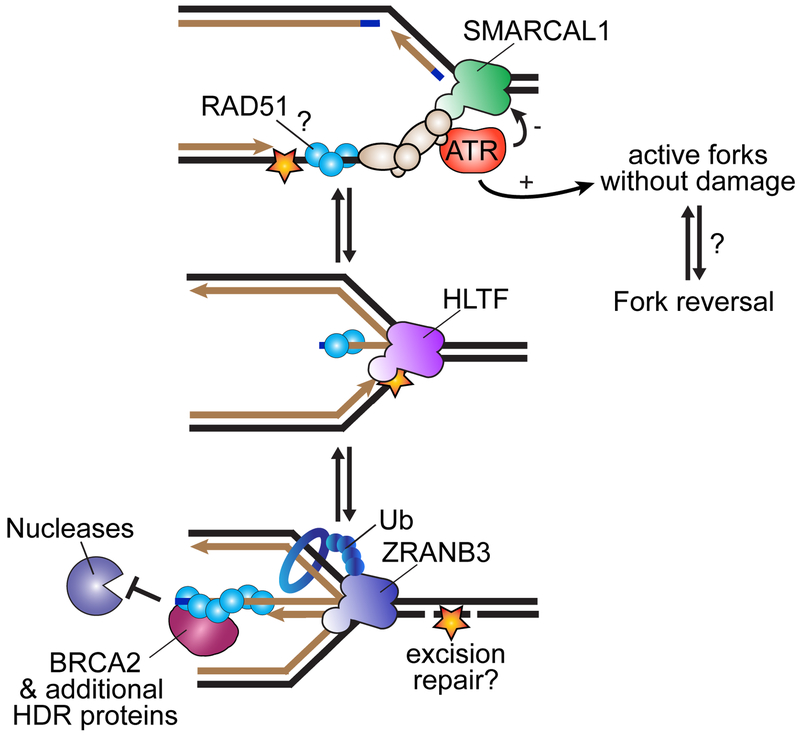
The replisome quickly and accurately copies billions of DNA bases each cell division cycle. However, it can make errors, especially when the template DNA is damaged. In these cases, replication-coupled repair mechanisms remove the mistake or repair the template lesions to ensure high fidelity and complete copying of the genome. Failures in these genome maintenance activities generate mutations, rearrangements, and chromosome segregation problems that cause many human diseases. In this review, I provide a broad overview of replication-coupled repair pathways, explaining how they fix polymerase mistakes, respond to template damage that acts as obstacles to the replisome, deal with broken forks, and impact human health and disease.
Keywords: DNA damage; DNA replication; break-induced replication; cancer; crosslink repair; excision repair; fork protection; fork reversal; mismatch repair; replication stress.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
The authors declare no competing interests. eTOC blurb: Replication-coupled DNA repair minimizes replication errors and prevents disease. This review discusses these mechanisms and provides perspective on important concepts and outstanding questions.
Figures




References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
