

Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy
- PMID: 31174843
- PMCID: PMC6719339
- DOI: 10.1016/j.trecan.2019.03.005
Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy
Abstract
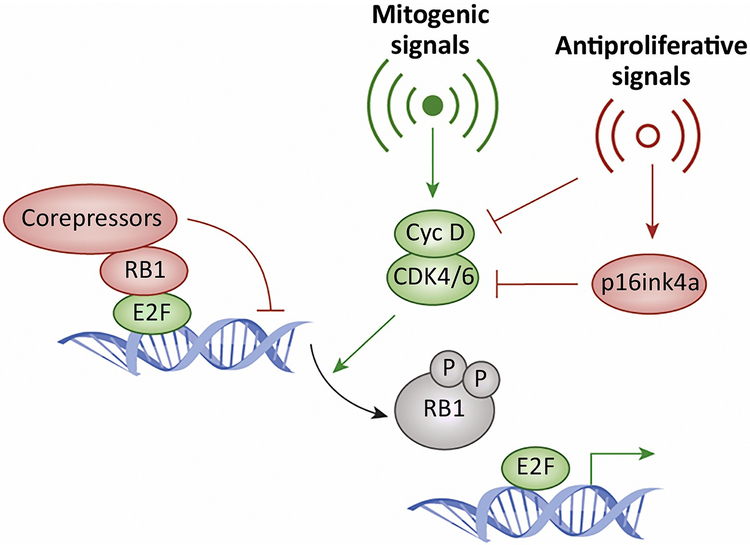
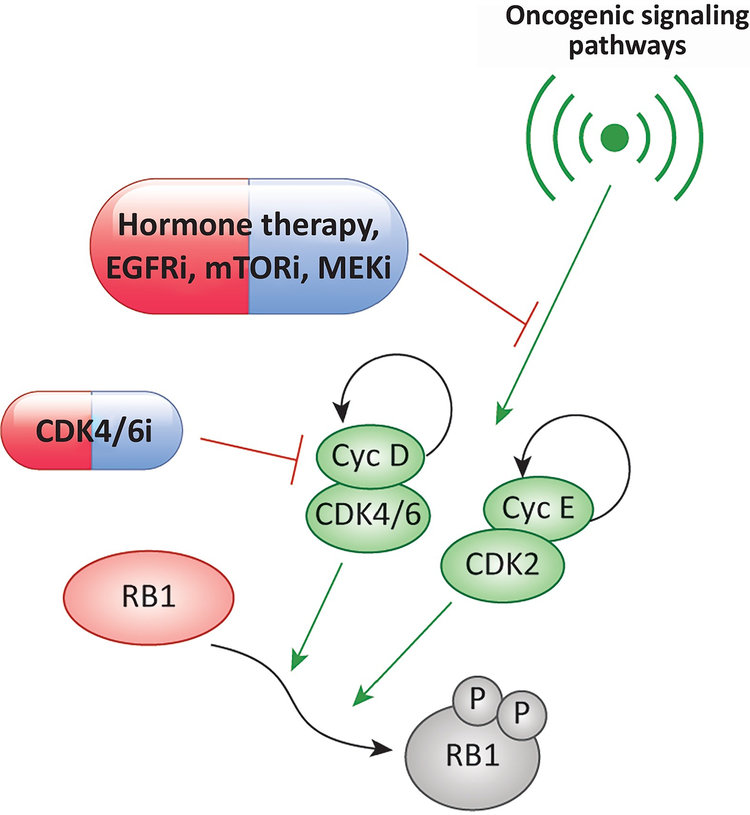
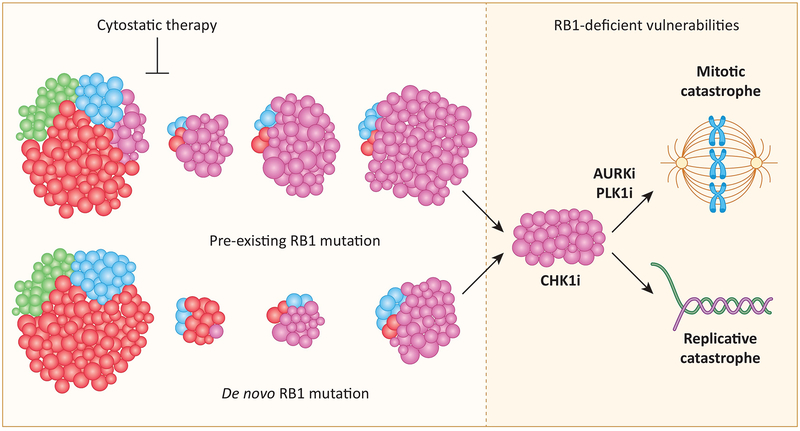
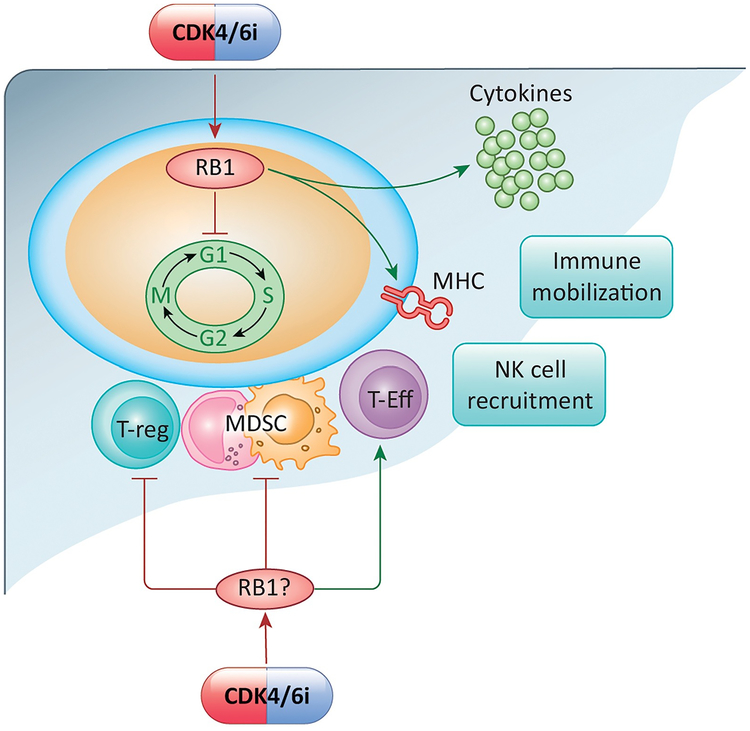
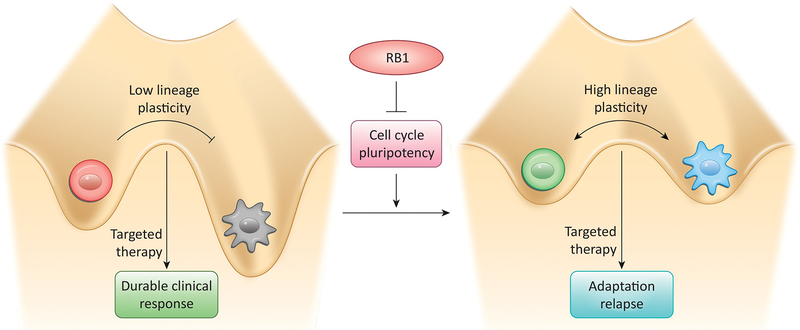
Recent studies highlight the importance of the RB1 tumor suppressor as a target for cancer therapy. Canonically, RB1 regulates cell cycle progression and represents the downstream target for cyclin-dependent kinase (CDK) 4/6 inhibitors that are in clinical use. However, newly discovered features of the RB1 pathway suggest new therapeutic strategies to counter resistance and improve precision medicine. These therapeutic strategies include deepening cell cycle exit with CDK4/6 inhibitor combinations, selectively targeting tumors that have lost RB1, and expanding therapeutic index by mitigating therapy-associated adverse effects. In addition, RB1 impacts immunological features of tumors and the microenvironment that can enhance sensitivity to immunotherapy. Lastly, RB1 specifies epigenetically determined cell lineage states that are disrupted during therapy resistance and could be re-installed through the direct use of epigenetic therapies. Thus, new opportunities are emerging to improve cancer therapy by exploiting the RB1 pathway.
Keywords: CDK4; E2F; EZH2; RB1; cyclin; epigenetics; immunotherapy; palbociclib; retinoblastoma.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
