

Management of bone disease in cystinosis: Statement from an international conference
- PMID: 31177550
- PMCID: PMC7379238
- DOI: 10.1002/jimd.12134
Management of bone disease in cystinosis: Statement from an international conference
Abstract
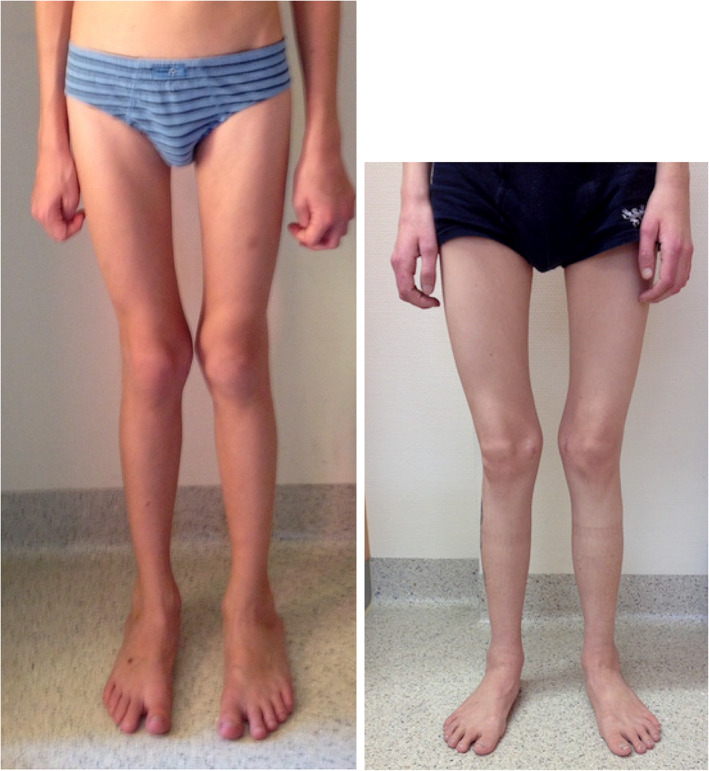
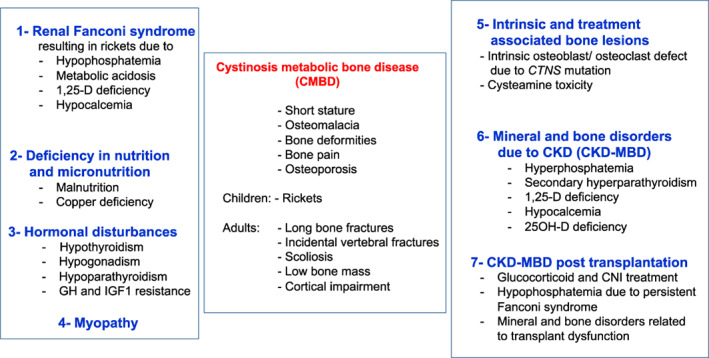
Cystinosis is an autosomal recessive storage disease due to impaired transport of cystine out of lysosomes. Since the accumulation of intracellular cystine affects all organs and tissues, the management of cystinosis requires a specialized multidisciplinary team consisting of pediatricians, nephrologists, nutritionists, ophthalmologists, endocrinologists, neurologists' geneticists, and orthopedic surgeons. Treatment with cysteamine can delay or prevent most clinical manifestations of cystinosis, except the renal Fanconi syndrome. Virtually all individuals with classical, nephropathic cystinosis suffer from cystinosis metabolic bone disease (CMBD), related to the renal Fanconi syndrome in infancy and progressive chronic kidney disease (CKD) later in life. Manifestations of CMBD include hypophosphatemic rickets in infancy, and renal osteodystrophy associated with CKD resulting in bone deformities, osteomalacia, osteoporosis, fractures, and short stature. Assessment of CMBD involves monitoring growth, leg deformities, blood levels of phosphate, electrolytes, bicarbonate, calcium, and alkaline phosphatase, periodically obtaining bone radiographs, determining levels of critical hormones and vitamins, such as thyroid hormone, parathyroid hormone, 25(OH) vitamin D, and testosterone in males, and surveillance for nonrenal complications of cystinosis such as myopathy. Treatment includes replacement of urinary losses, cystine depletion with oral cysteamine, vitamin D, hormone replacement, physical therapy, and corrective orthopedic surgery. The recommendations in this article came from an expert meeting on CMBD that took place in Salzburg, Austria, in December 2016.
Keywords: CKD-MBD; Fanconi syndrome; chronic kidney disease; cystinosis; cystinosis metabolic bone disease; hypophosphatemic rickets; transplantation.
© 2019 The Authors. Journal of Inherited Metabolic Disease published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
Atif Awan, Justine Bacchetta, Frank Rauch, Erik Harms, Bernd Hoppe, Nadine Herzig, Bernd Hoppe, Ewa Elenberg, William A. Gahl, Christian Koeppl, Elena Levtchenko, Malcolm Lewis, Galina Nesterova, Fernando Santos, Karl P. Schlingmann, Aude Servais, Neveen A. Soliman, Guentehr Steidle, Rezan Topaloglu, Ulrike Treikauskas, Alexey Tsygin, Koenraad Veys, Josef Zustin, Rodo v. Vigier have no conflict of interest. Gema Ariceta received speaker honorarium from Chiesi, Orphan and Horizon and consulting fee from Chiesi. Susanne Bechtold received speaker honorarium from Sandoz and consulting fee from Alexion. Carsten Bergmann received speaker honorarium from Alexion. George Deschennes received consulting honorarium from Chiesi. Dieter Haffner received speaker honorarium from Horizon, Chiesi and Orphan. Katharina Hohenfellner received speaker fee from Orphan and consulting honorarium from Leadiant. This article does not contain any studies with human or animal subjects performed by the any of the authors.
Figures
References
-
- Gahl WA, Bashan N, Tietze F, Bernardini I, Schulman JD. Lysosomal cystine transport is defective in cystinosis. Science. 1982;217:1263‐1265. - PubMed
-
- Town M, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319‐324. - PubMed
-
- Nesterova G, Gahl WA. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews(®) [Internet]. Seattle (WA): University of Washington, Seattle; 2001:1993‐2017.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
