

Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain
- PMID: 31177989
- PMCID: PMC6556959
- DOI: 10.1186/s40478-019-0747-x
Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain
Erratum in
-
Publisher Correction to: Acta Neuropathologica Communications, volume 7.Acta Neuropathol Commun. 2019 Aug 14;7(1):131. doi: 10.1186/s40478-019-0784-5. Acta Neuropathol Commun. 2019. PMID: 31412936 Free PMC article.
Abstract
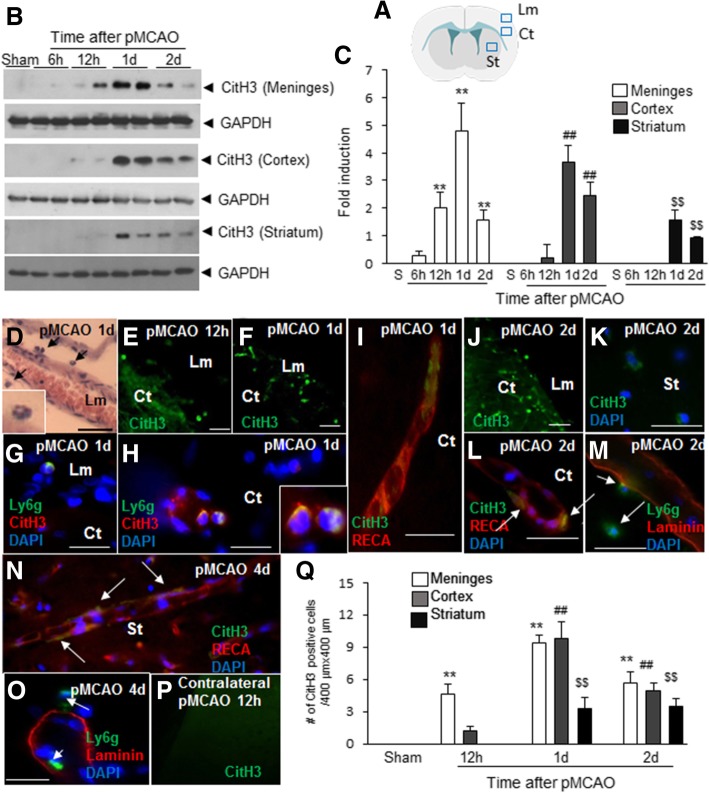
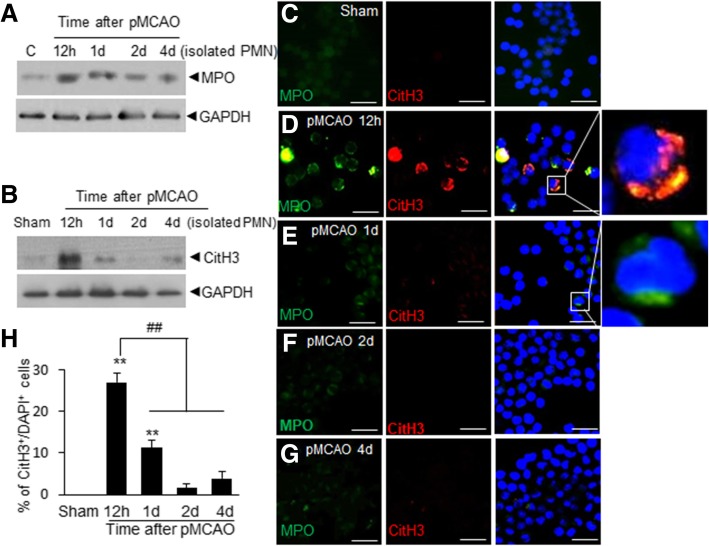
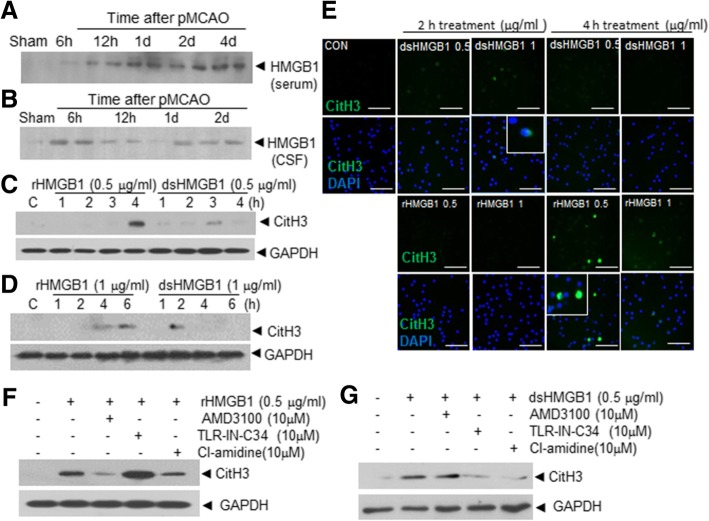
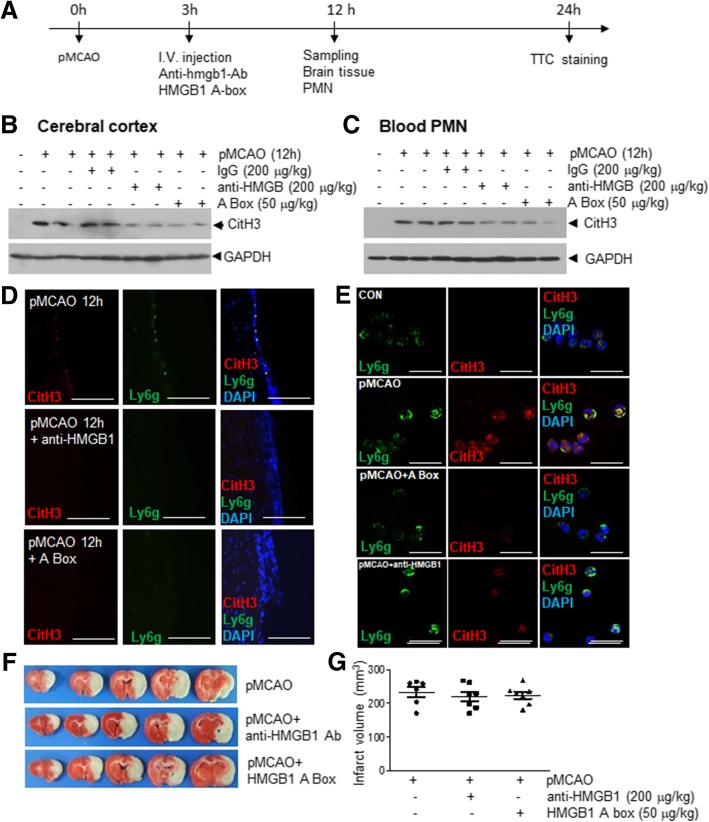
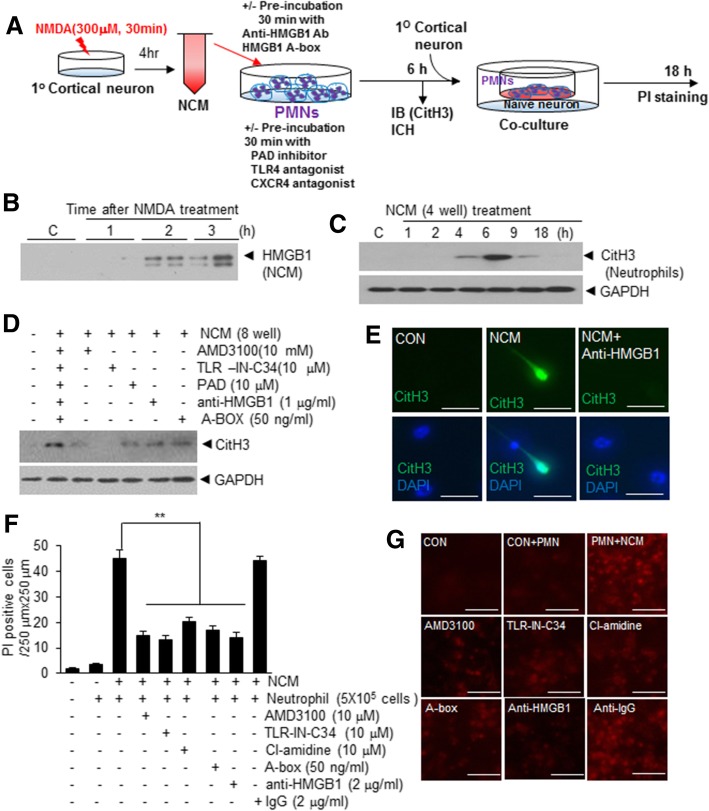
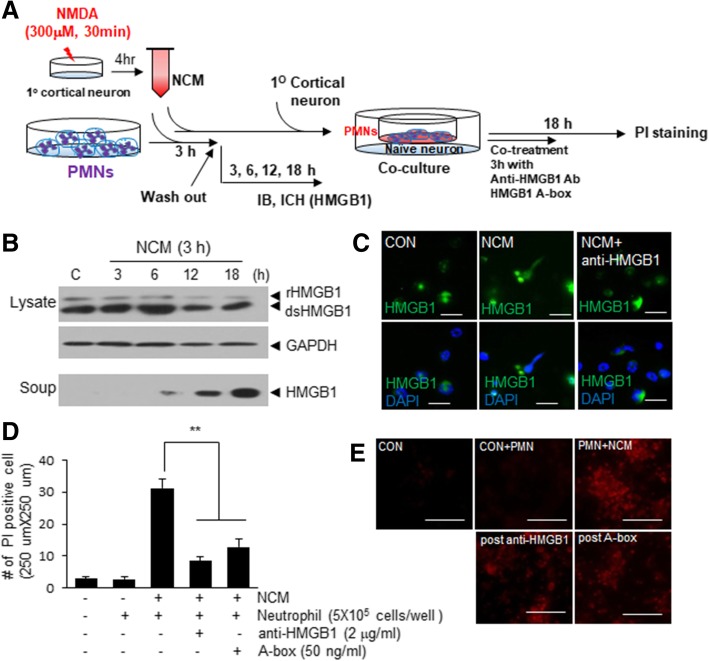
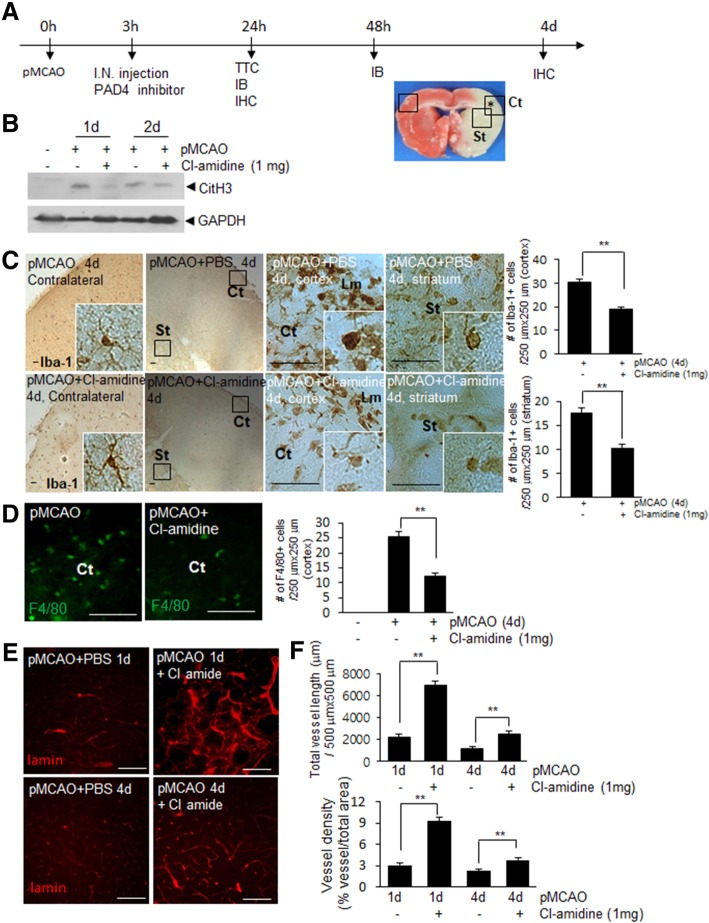
It has been reported that neutrophil extracellular traps (NETs) play important roles in non-infectious diseases. In ischemic stroke, neutrophils infiltrate damaged brain tissue soon after injury and aggravate inflammation. Using a rat permanent MCAO model, we showed citrullinated histone H3+ (CitH3, a marker of NETosis) induction in neutrophils in leptomeninges and in peripheral blood soon after MCAO. Entry of CitH3+ cells occurred through leptomeninges after 6 h of MCAO and these cells were observed in cerebral cortex from 12 h and subsequently in striatum. It is interesting to note that CitH3+ induction began in circulating neutrophils before they migrated to brain parenchyma and they were detected as intact or lysed form. High mobility group box 1 (HMGB1), a danger associated molecular pattern (DAMP) molecule, was accumulated massively in serum after permanent MCAO and plays a critical role in CitH3 inductions in neutrophils in brain parenchyma and in peripheral blood. Both the all-thiol and disulfide types of HMGB1 induced CitH3 via their specific receptors, CXCR4 and TLR4, respectively. Importantly, HMGB1 not only induced NETosis but was included as a part of the extruded NETs, and contribute to NETosis-mediated neuronal death. Therefore, it would appear a vicious cycle exists between neuronal cell death and NETosis and HMGB1 mediates detrimental effects exerted by this cycle. When NETosis was suppressed by a PAD inhibitor in MCAO animals, delayed immune cell infiltrations were markedly suppressed and damages in blood vessels were significantly mitigated. The study shows NETosis with the involvement of HMGB1 as a mediator in a vicious cycle aggravates inflammation and subsequent damage in the ischemic brain.
Keywords: HMGB1; Inflammation; MCAO; NETosis; Permanent ischemia.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
