

AKR1C1 controls cisplatin-resistance in head and neck squamous cell carcinoma through cross-talk with the STAT1/3 signaling pathway
- PMID: 31182137
- PMCID: PMC6558898
- DOI: 10.1186/s13046-019-1256-2
AKR1C1 controls cisplatin-resistance in head and neck squamous cell carcinoma through cross-talk with the STAT1/3 signaling pathway
Abstract
Background: Cisplatin is the first-line chemotherapy used against most upper aerodigestive tract carcinomas. In head and neck cancer, sensitivity to cisplatin remains the key issue in treatment response and outcome. Genetic heterogeneity and aberrant gene expression may be the intrinsic factors that cause primary cisplatin-resistance.
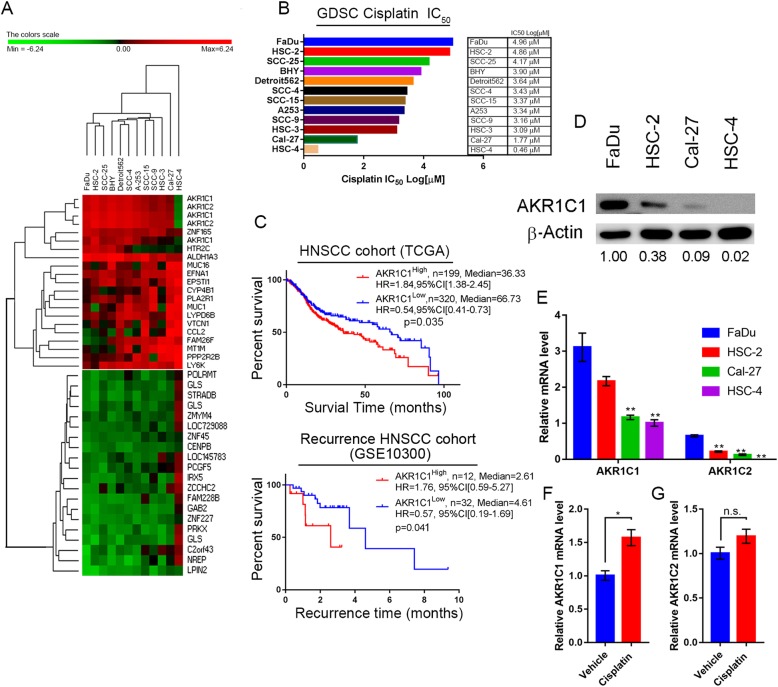
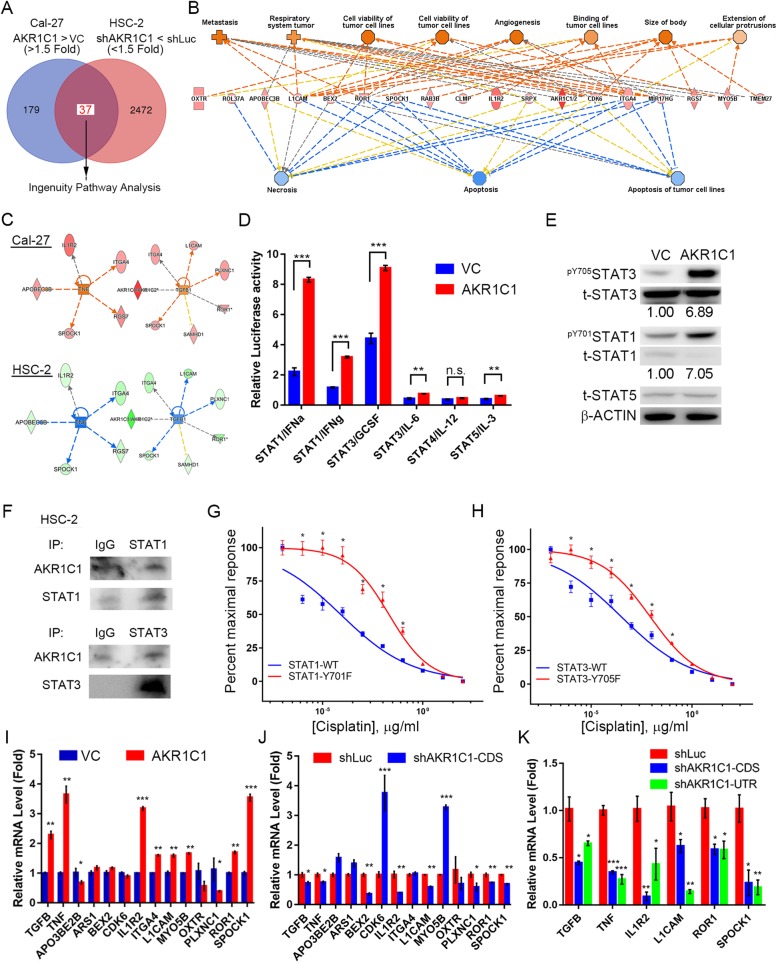
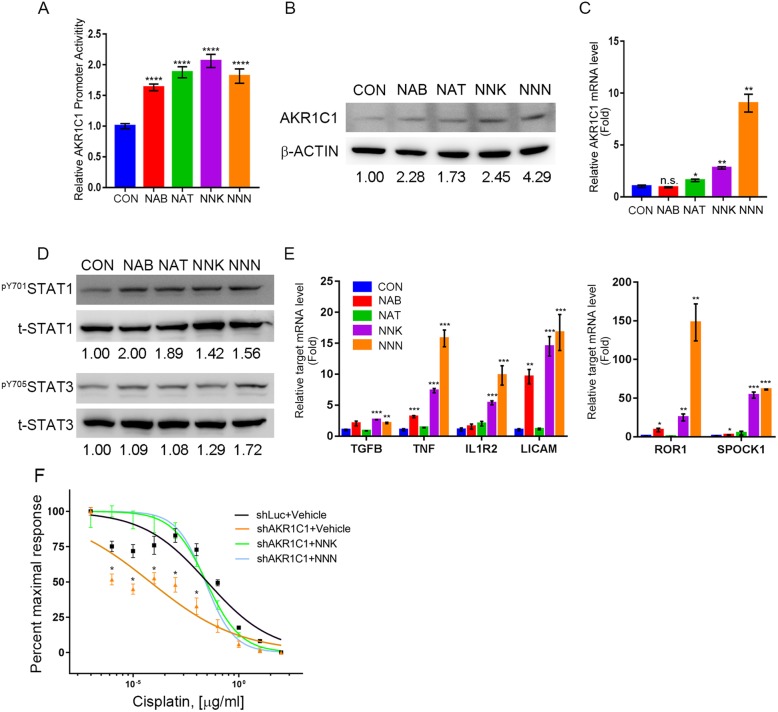
Methods: Combination of the HNSCC gene expression data and the cisplatin sensitivity results from public database. We found that aldo-keto reductase family 1 member C1 (AKR1C1) may be associated with cisplatin sensitivity in HNSCC treatment of naïve cells. We examined the AKR1C1 expression and its correlation with cisplatin IC50 and prognosis in patients. The in vitro and in vivo AKR1C1 functions in cisplatin-resistance through overexpression or knockdown assays, respectively. cDNA microarrays were used to identify the upstream regulators that modulate AKR1C1-induced signaling in HNSCC. Finally, we used the cigarette metabolites to promote AKR1C1 expression and ruxolitinib to overcome AKR1C1-induced cisplatin-resistance.
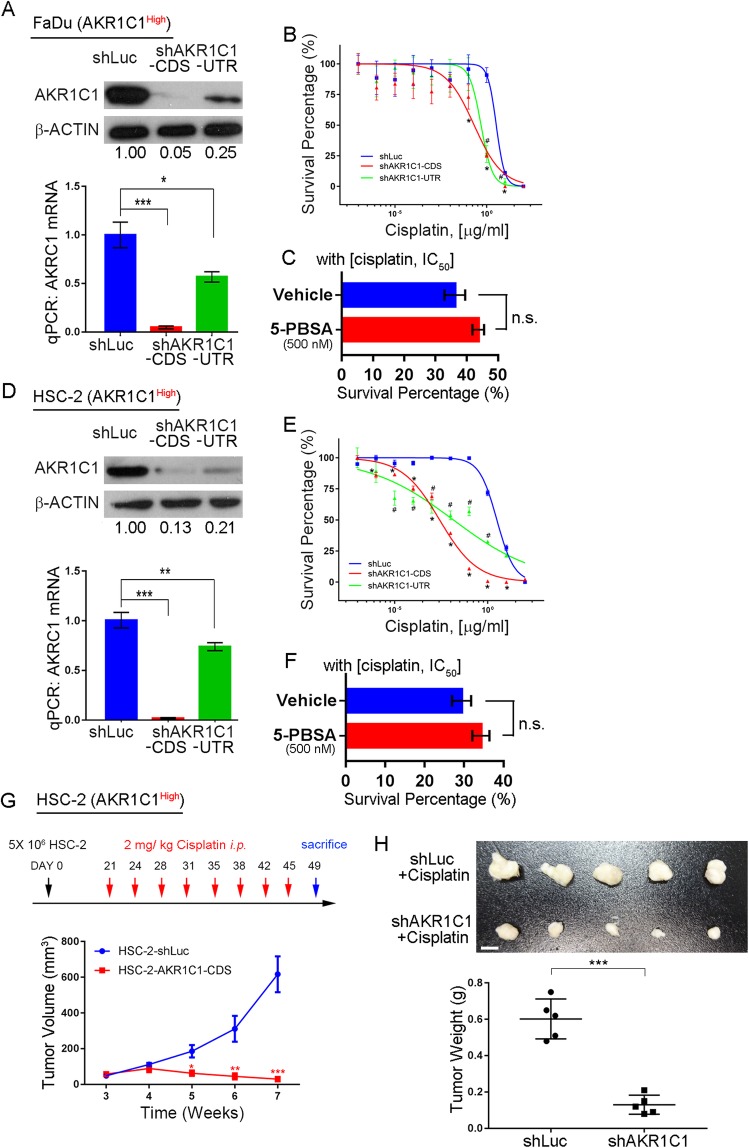
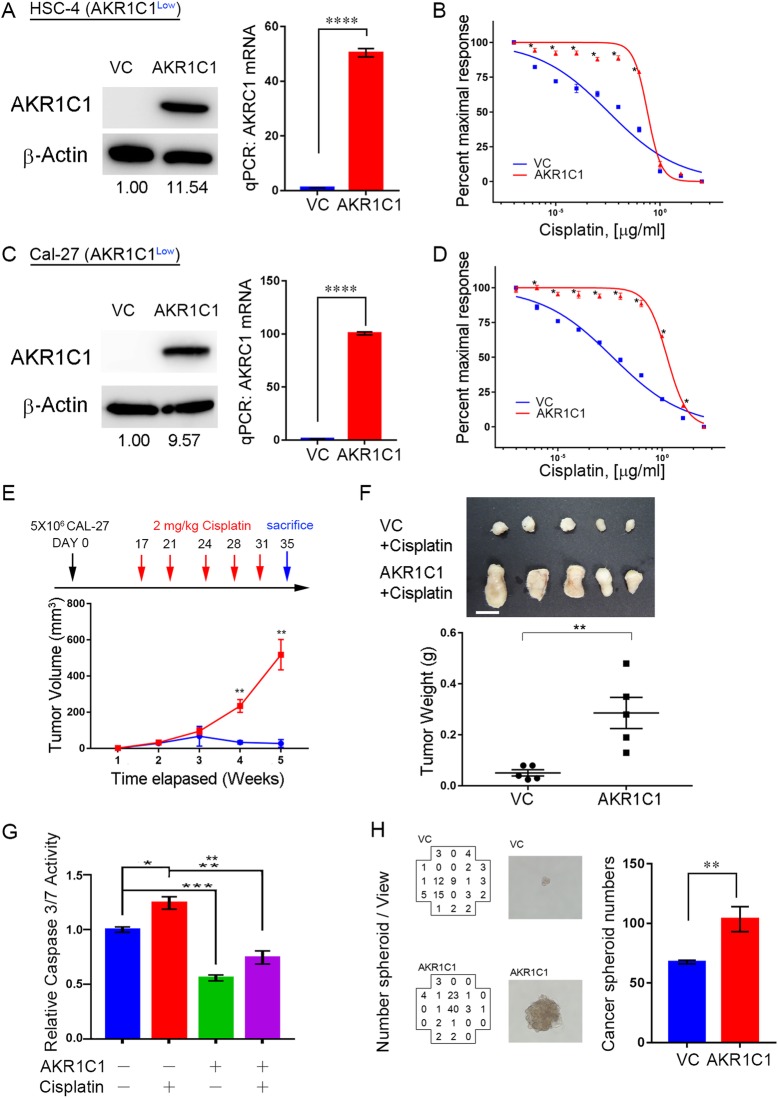
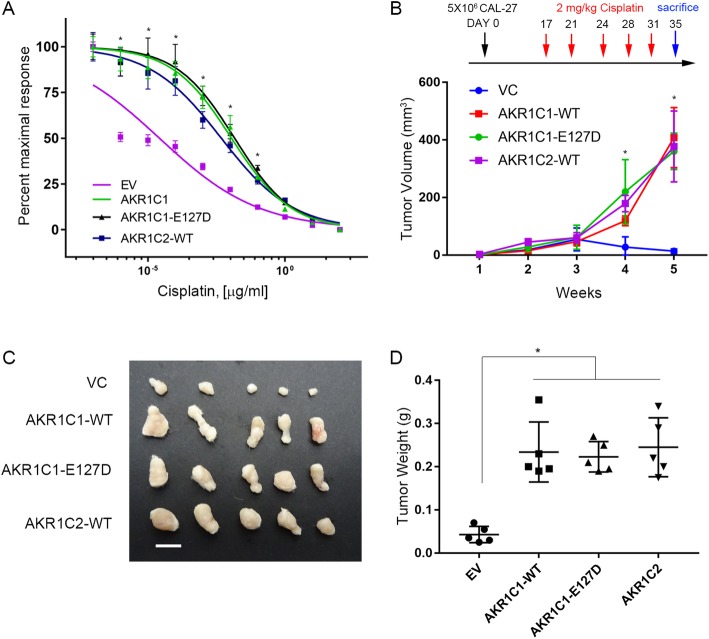
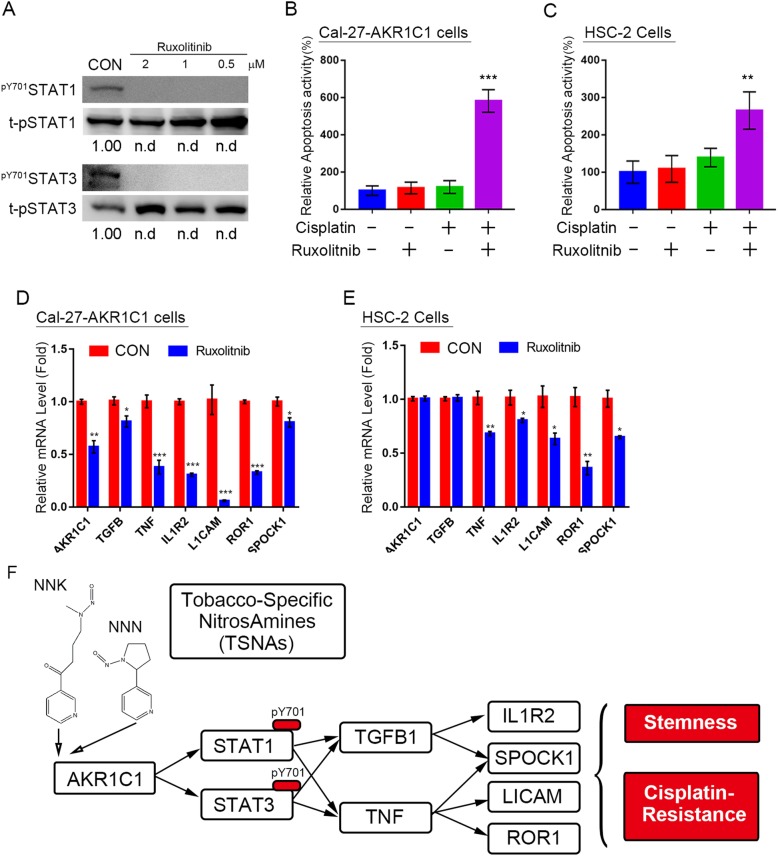
Results: AKR1C1 positively correlates to cisplatin-resistance in HNSCC cells. AKR1C1 is a poor prognostic factor for recurrence and death of HNSCC patients. Silencing of AKR1C1 not only reduced in vitro IC50 but also increased in vivo cisplatin responses and vise versa in overexpression cells. Cigarette metabolites also promote AKR1C1 expression. Transcriptome analyses revealed that STAT1 and STAT3 activation enable AKR1C1-induced cisplatin-resistance and can be overcome by ruxolitinib treatment.
Conclusions: AKR1C1 is a crucial regulator for cisplatin-resistance in HNSCC and also poor prognostic marker for patients. Targeting the AKR1C1-STAT axis may provide a new therapeutic strategy to treat patients who are refractory to cisplatin treatment.
Keywords: AKR1C1; Cisplatin-resistance; HNSCC; Ruxolitinib; STATs.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
