

Raising fluid walls around living cells
- PMID: 31183401
- PMCID: PMC6551168
- DOI: 10.1126/sciadv.aav8002
Raising fluid walls around living cells
Abstract
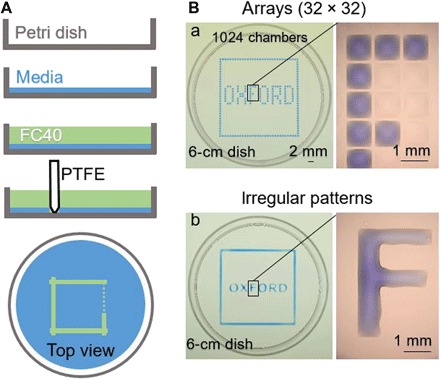
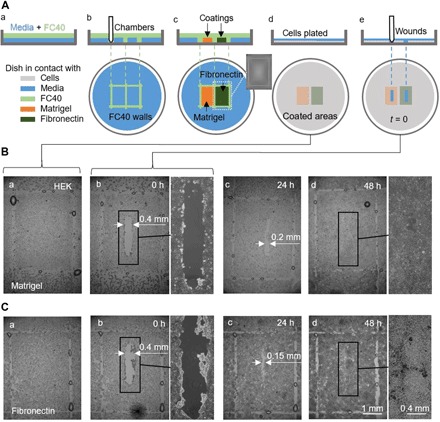
An effective transformation of the cell culture dishes that biologists use every day into microfluidic devices would open many avenues for miniaturizing cell-based workflows. In this article, we report a simple method for creating microfluidic arrangements around cells already growing on the surface of standard petri dishes, using the interface between immiscible fluids as a "building material." Conventional dishes are repurposed into sophisticated microfluidic devices by reshaping, on demand, the fluid structures around living cells. Moreover, these microfluidic arrangements can be further reconfigured during experiments, which is impossible with most existing microfluidic platforms. The method is demonstrated using workflows involving cell cloning, the selection of a particular clone from among others in a dish, drug treatments, and wound healing. The versatility of the approach and its biologically friendly aspects may hasten uptake by biologists of microfluidics, so the technology finally fulfills its potential.
Figures




References
-
- J. Berthier, K. A. Brakke, The Physics of Microdroplets (John Wiley & Sons, 2012).
-
- Sackmann E. K., Fulton A. L., Beebe D. J., The present and future role of microfluidics in biomedical research. Nature 507, 181–189 (2014). - PubMed
-
- Berthier E., Young E. W. K., Beebe D., Engineers are from PDMS-land, biologists are from Polystyrenia. Lab Chip 12, 1224–1237 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
