

The Role of HMGB1, a Nuclear Damage-Associated Molecular Pattern Molecule, in the Pathogenesis of Lung Diseases
- PMID: 31184204
- PMCID: PMC6765066
- DOI: 10.1089/ars.2019.7818
The Role of HMGB1, a Nuclear Damage-Associated Molecular Pattern Molecule, in the Pathogenesis of Lung Diseases
Abstract
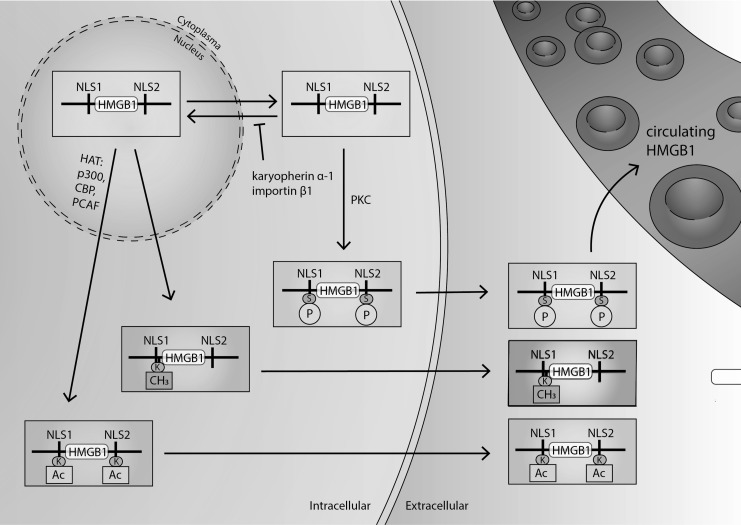
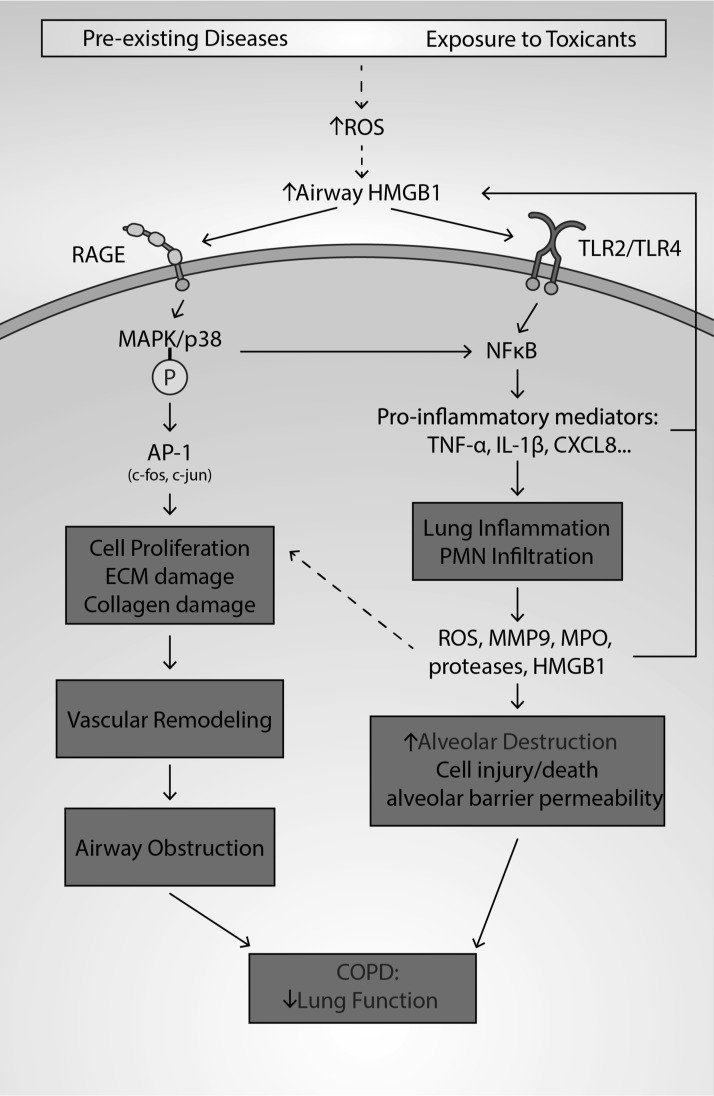
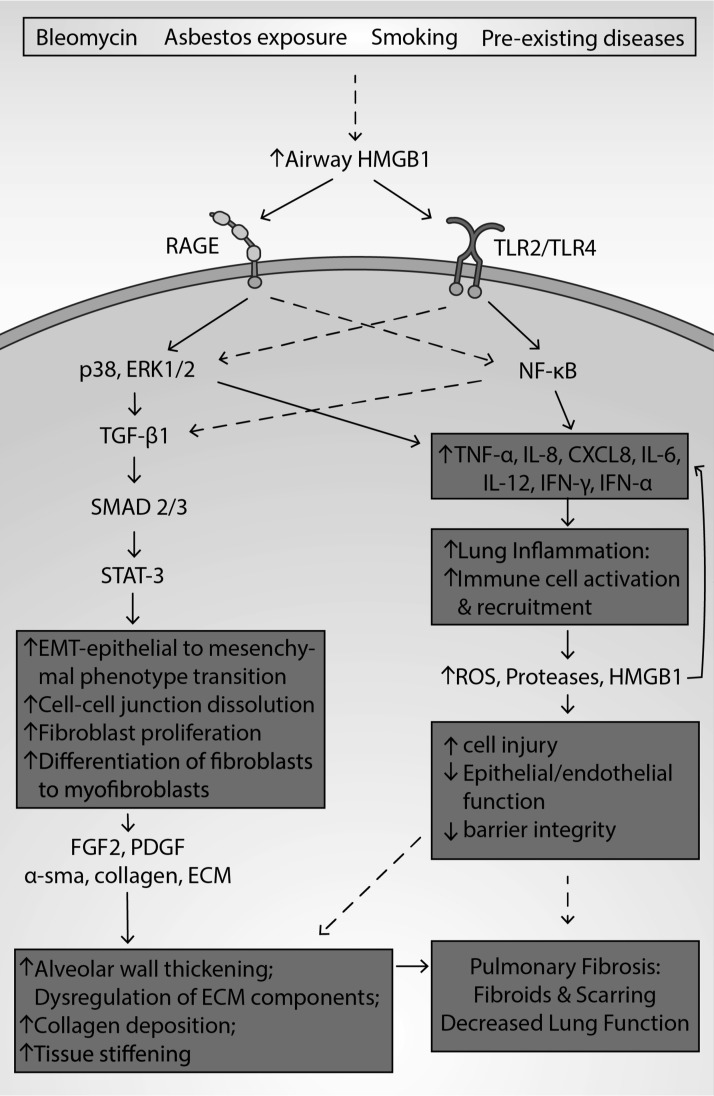
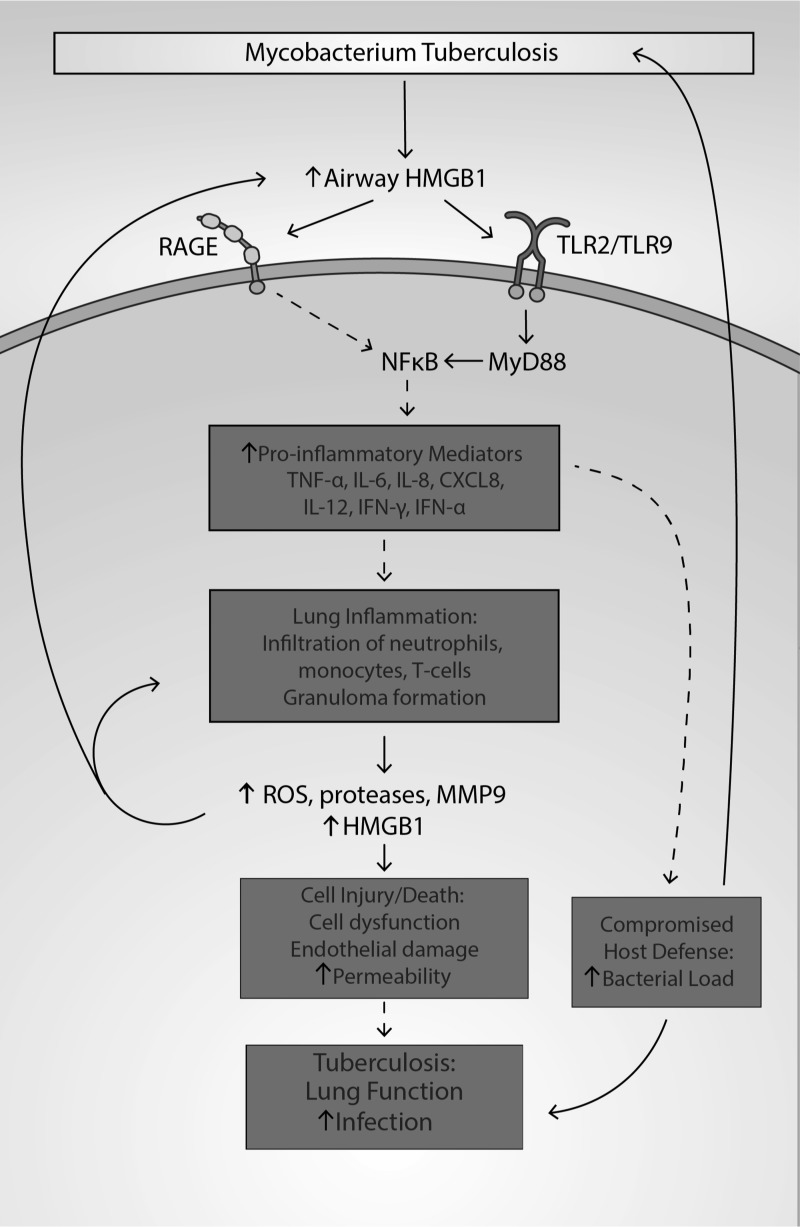
Significance: High-mobility group protein box 1 (HMGB1), a ubiquitous nuclear protein, regulates chromatin structure and modulates the expression of many genes involved in the pathogenesis of lung cancer and many other lung diseases, including those that regulate cell cycle control, cell death, and DNA replication and repair. Extracellular HMGB1, whether passively released or actively secreted, is a danger signal that elicits proinflammatory responses, impairs macrophage phagocytosis and efferocytosis, and alters vascular remodeling. This can result in excessive pulmonary inflammation and compromised host defense against lung infections, causing a deleterious feedback cycle. Recent Advances: HMGB1 has been identified as a biomarker and mediator of the pathogenesis of numerous lung disorders. In addition, post-translational modifications of HMGB1, including acetylation, phosphorylation, and oxidation, have been postulated to affect its localization and physiological and pathophysiological effects, such as the initiation and progression of lung diseases. Critical Issues: The molecular mechanisms underlying how HMGB1 drives the pathogenesis of different lung diseases and novel therapeutic approaches targeting HMGB1 remain to be elucidated. Future Directions: Additional research is needed to identify the roles and functions of modified HMGB1 produced by different post-translational modifications and their significance in the pathogenesis of lung diseases. Such studies will provide information for novel approaches targeting HMGB1 as a treatment for lung diseases.
Keywords: HMGB1; cancer; fibrosis; inflammatory lung injury; pulmonary infection; pulmonary vascular remodeling.
Conflict of interest statement
The authors declare no competing financial interest.
Figures









References
-
- Aaron SD, Angel JB, Lunau M, Wright K, Fex C, Le Saux N, and Dales RE. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 163: 349–355, 2001 - PubMed
-
- Abeyama K, Stern DM, Ito Y, Kawahara K, Yoshimoto Y, Tanaka M, Uchimura T, Ida N, Yamazaki Y, Yamada S, Yamamoto Y, Yamamoto H, Iino S, Taniguchi N, and Maruyama I. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest 115: 1267–1274, 2005 - PMC - PubMed
-
- Abraham E, Arcaroli J, Carmody A, Wang H, and Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol 1950 165: 2950–2954, 2000 - PubMed
-
- Agustí AGN, Roca J, Gea J, Wagner PD, Xaubet A, and Rodriguez-Roisin R. Mechanisms of gas-exchange impairment in idiopathic pulmonary fibrosis. Am Rev Respir Dis 143: 219–225, 1991 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
