

DNA methylation profiling allows for characterization of atrial and ventricular cardiac tissues and hiPSC-CMs
- PMID: 31186048
- PMCID: PMC6560887
- DOI: 10.1186/s13148-019-0679-0
DNA methylation profiling allows for characterization of atrial and ventricular cardiac tissues and hiPSC-CMs
Abstract
Background: Cardiac disease modelling using human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM) requires thorough insight into cardiac cell type differentiation processes. However, current methods to discriminate different cardiac cell types are mostly time-consuming, are costly and often provide imprecise phenotypic evaluation. DNA methylation plays a critical role during early heart development and cardiac cellular specification. We therefore investigated the DNA methylation pattern in different cardiac tissues to identify CpG loci for further cardiac cell type characterization.
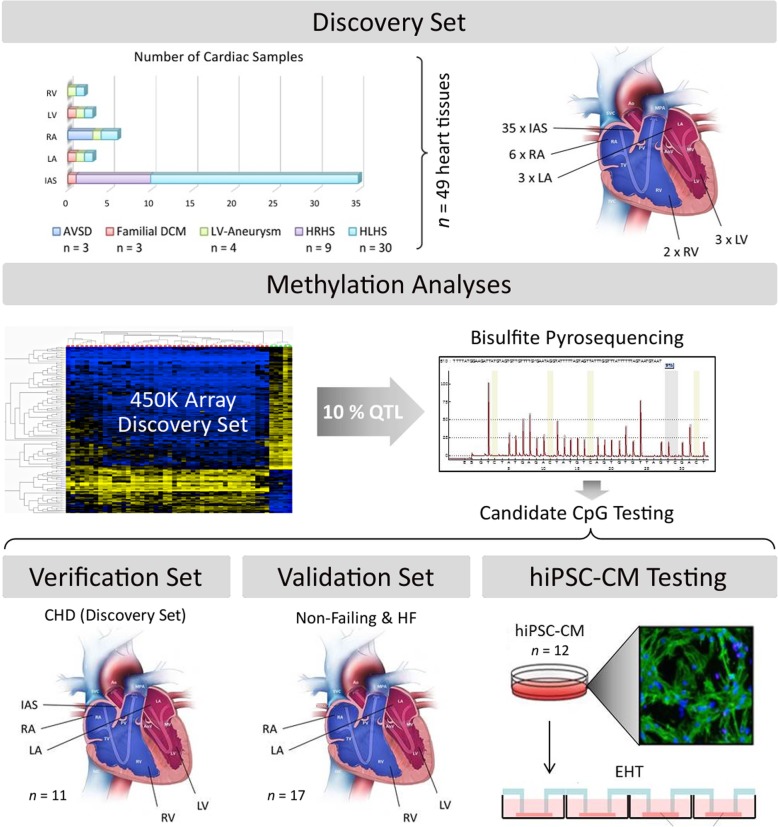
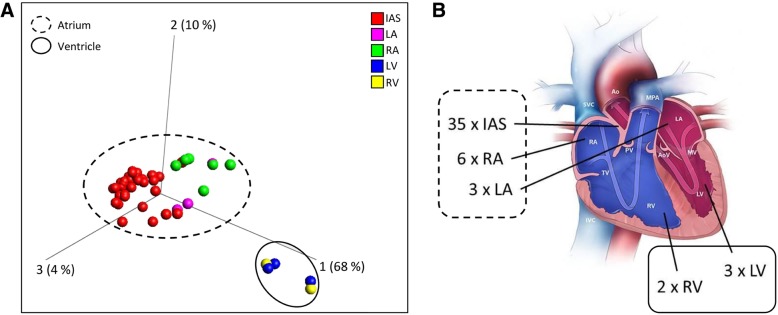
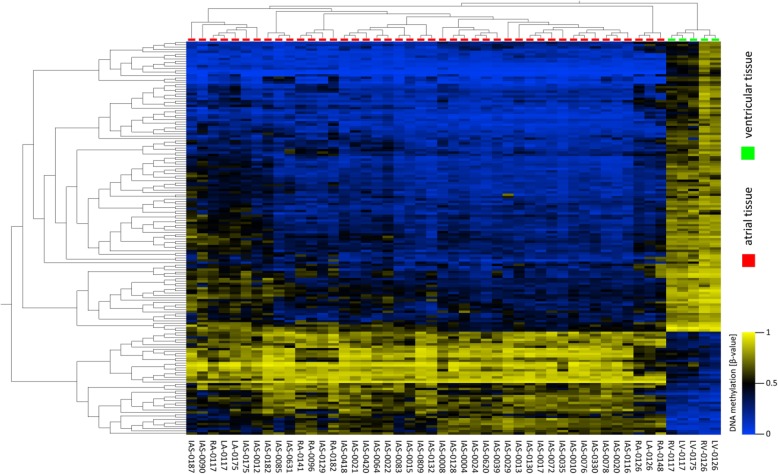
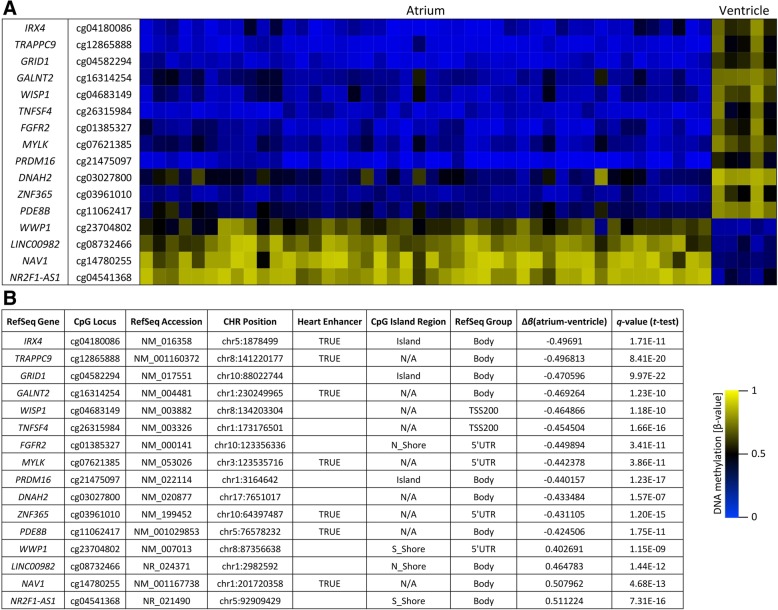
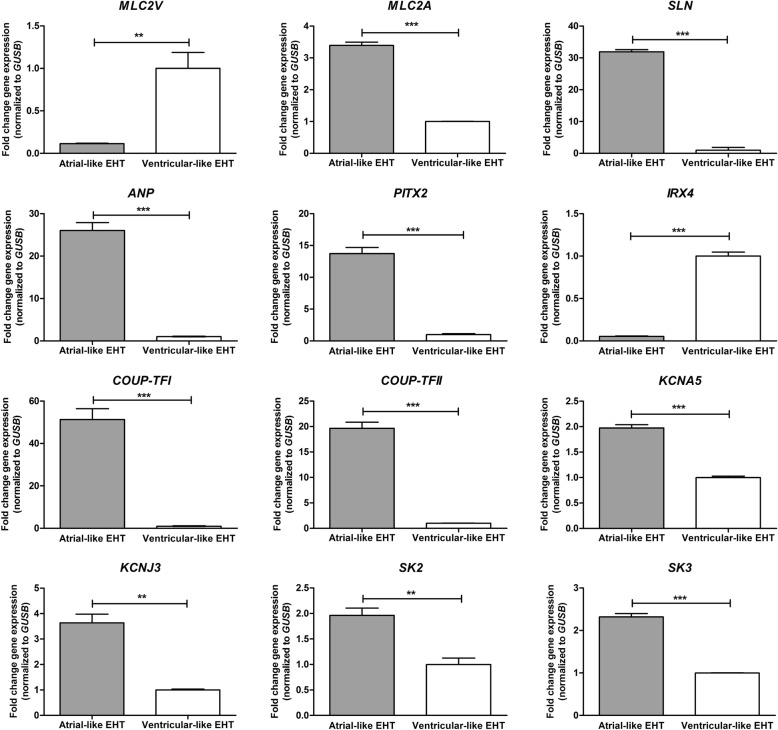
Results: An array-based genome-wide DNA methylation analysis using Illumina Infinium HumanMethylation450 BeadChips led to the identification of 168 differentially methylated CpG loci in atrial and ventricular human heart tissue samples (n = 49) from different patients with congenital heart defects (CHD). Systematic evaluation of atrial-ventricular DNA methylation pattern in cardiac tissues in an independent sample cohort of non-failing donor hearts and cardiac patients using bisulfite pyrosequencing helped us to define a subset of 16 differentially methylated CpG loci enabling precise characterization of human atrial and ventricular cardiac tissue samples. This defined set of reproducible cardiac tissue-specific DNA methylation sites allowed us to consistently detect the cellular identity of hiPSC-CM subtypes.
Conclusion: Testing DNA methylation of only a small set of defined CpG sites thus makes it possible to distinguish atrial and ventricular cardiac tissues and cardiac atrial and ventricular subtypes of hiPSC-CMs. This method represents a rapid and reliable system for phenotypic characterization of in vitro-generated cardiomyocytes and opens new opportunities for cardiovascular research and patient-specific therapy.
Keywords: 450K array; Bisulfite pyrosequencing; Cardiac tissue-specific DNA methylation; DNA methylation; Engineered heart tissue (EHT); Human induced pluripotent stem cell derived cardiomyocytes (hiPSC-CM).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


Similar articles
-
Engineered heart tissue models from hiPSC-derived cardiomyocytes and cardiac ECM for disease modeling and drug testing applications.Acta Biomater. 2019 Jul 1;92:145-159. doi: 10.1016/j.actbio.2019.05.016. Epub 2019 May 7. Acta Biomater. 2019. PMID: 31075518
-
Characterization of Ventricular and Atrial Cardiomyocyte Subtypes from Human-Induced Pluripotent Stem Cells.Methods Mol Biol. 2021;2320:135-149. doi: 10.1007/978-1-0716-1484-6_14. Methods Mol Biol. 2021. PMID: 34302655
-
Single-Cell Transcriptomics of Engineered Cardiac Tissues From Patient-Specific Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveals Abnormal Developmental Trajectory and Intrinsic Contractile Defects in Hypoplastic Right Heart Syndrome.J Am Heart Assoc. 2020 Oct 20;9(20):e016528. doi: 10.1161/JAHA.120.016528. Epub 2020 Oct 16. J Am Heart Assoc. 2020. PMID: 33059525 Free PMC article.
-
Human-induced pluripotent stem cell-derived cardiomyocytes, 3D cardiac structures, and heart-on-a-chip as tools for drug research.Pflugers Arch. 2021 Jul;473(7):1061-1085. doi: 10.1007/s00424-021-02536-z. Epub 2021 Feb 24. Pflugers Arch. 2021. PMID: 33629131 Free PMC article. Review.
-
Modeling Cardiovascular Diseases with hiPSC-Derived Cardiomyocytes in 2D and 3D Cultures.Int J Mol Sci. 2020 May 11;21(9):3404. doi: 10.3390/ijms21093404. Int J Mol Sci. 2020. PMID: 32403456 Free PMC article. Review.
Cited by
-
TANC1 methylation as a novel biomarker for the diagnosis of patients with anti-tuberculosis drug-induced liver injury.Sci Rep. 2021 Aug 31;11(1):17423. doi: 10.1038/s41598-021-96869-5. Sci Rep. 2021. PMID: 34465797 Free PMC article.
-
Epigenetics and Heart Development.Front Cell Dev Biol. 2021 May 6;9:637996. doi: 10.3389/fcell.2021.637996. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34026751 Free PMC article. Review.
-
Causal Associations of DNA Methylation and Cardiovascular Disease: A Two-Sample Mendelian Randomization Study.Glob Heart. 2024 May 14;19(1):48. doi: 10.5334/gh.1324. eCollection 2024. Glob Heart. 2024. PMID: 38765775 Free PMC article.
-
DNA Methylation and Telomeres-Their Impact on the Occurrence of Atrial Fibrillation during Cardiac Aging.Int J Mol Sci. 2023 Oct 28;24(21):15699. doi: 10.3390/ijms242115699. Int J Mol Sci. 2023. PMID: 37958686 Free PMC article. Review.
-
Genome-wide methylation analysis reveals differentially methylated CpG sites and altered expression of heart development-associated genes in fetuses with cardiac defects.Exp Ther Med. 2021 Sep;22(3):1032. doi: 10.3892/etm.2021.10464. Epub 2021 Jul 18. Exp Ther Med. 2021. PMID: 34373718 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
