

Translatome analysis reveals altered serine and glycine metabolism in T-cell acute lymphoblastic leukemia cells
- PMID: 31186416
- PMCID: PMC6559966
- DOI: 10.1038/s41467-019-10508-2
Translatome analysis reveals altered serine and glycine metabolism in T-cell acute lymphoblastic leukemia cells
Abstract
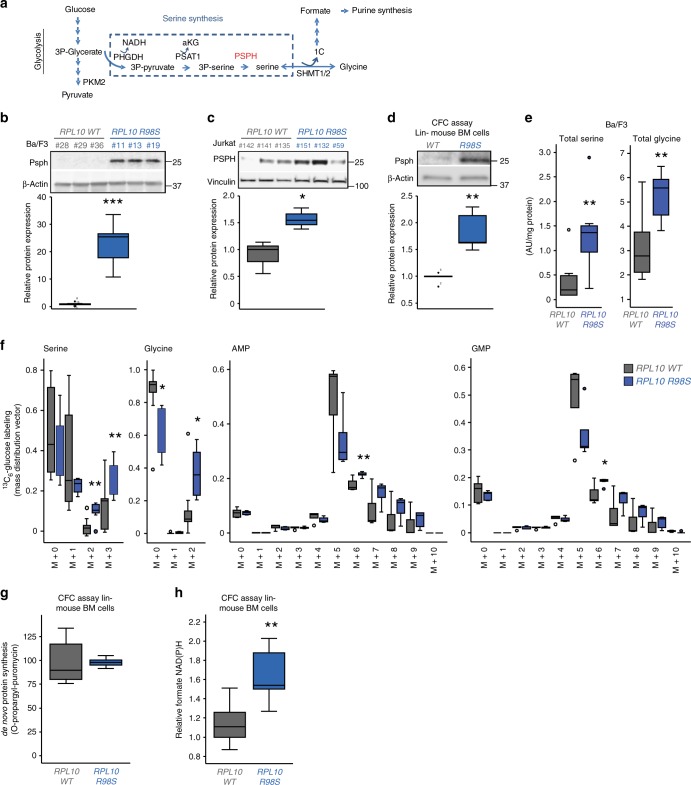
Somatic ribosomal protein mutations have recently been described in cancer, yet their impact on cellular transcription and translation remains poorly understood. Here, we integrate mRNA sequencing, ribosome footprinting, polysomal RNA sequencing and mass spectrometry datasets from a mouse lymphoid cell model to characterize the T-cell acute lymphoblastic leukemia (T-ALL) associated ribosomal RPL10 R98S mutation. Surprisingly, RPL10 R98S induces changes in protein levels primarily through transcriptional rather than translation efficiency changes. Phosphoserine phosphatase (PSPH), encoding a key serine biosynthesis enzyme, was the only gene with elevated transcription and translation leading to protein overexpression. PSPH upregulation is a general phenomenon in T-ALL patient samples, associated with elevated serine and glycine levels in xenograft mice. Reduction of PSPH expression suppresses proliferation of T-ALL cell lines and their capacity to expand in mice. We identify ribosomal mutation driven induction of serine biosynthesis and provide evidence supporting dependence of T-ALL cells on PSPH.
Conflict of interest statement
The authors declare no competing interests.
Figures







References
-
- Sulima SO, Hofman IJF, De Keersmaecker K, Dinman JD. How ribosomes translate cancer. Cancer Discov. 2017;7:1069–1087. doi: 10.1158/2159-8290.CD-17-0550. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 334946/EC | EC Seventh Framework Programm | FP7 Ideas: European Research Council (FP7-IDEAS-ERC - Specific Programme: "Ideas" Implementing the Seventh Framework Programme of the European Community for Research, Technological Development and Demonstration Activities (2007 to 2013))/International
- 2012-176/Stichting Tegen Kanker (Belgian Foundation Against Cancer)/International
- 2016-775/Stichting Tegen Kanker (Belgian Foundation Against Cancer)/International
- 2016-801/Stichting Tegen Kanker (Belgian Foundation Against Cancer)/International
- C14/18/104/Onderzoeksraad, KU Leuven (Research Council, KU Leuven)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
