

Mass spectra alignment using virtual lock-masses
- PMID: 31186508
- PMCID: PMC6560045
- DOI: 10.1038/s41598-019-44923-8
Mass spectra alignment using virtual lock-masses
Abstract
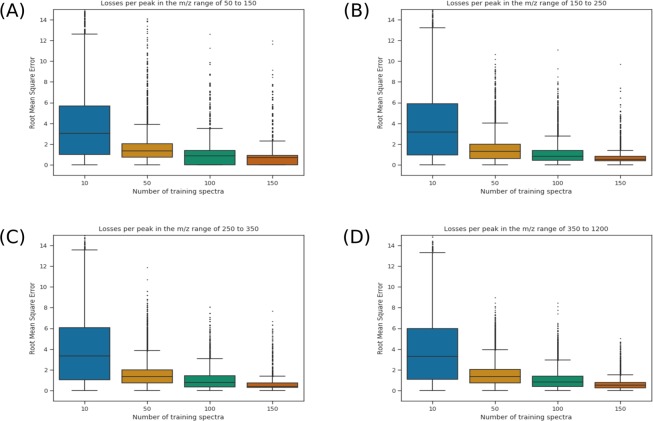
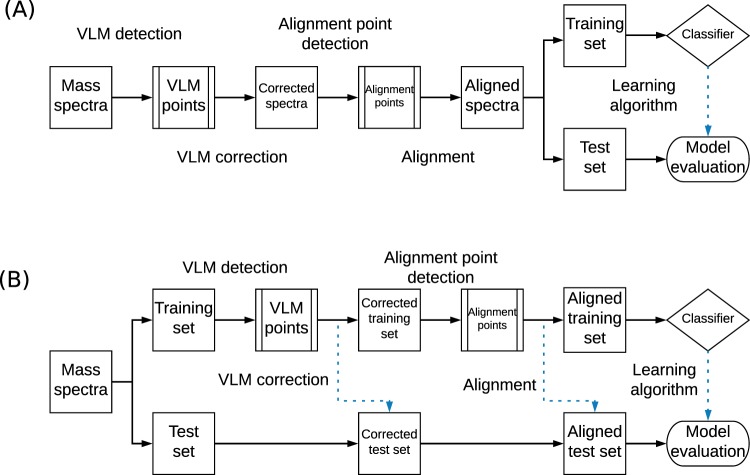
Mass spectrometry is a valued method to evaluate the metabolomics content of a biological sample. The recent advent of rapid ionization technologies such as Laser Diode Thermal Desorption (LDTD) and Direct Analysis in Real Time (DART) has rendered high-throughput mass spectrometry possible. It is used for large-scale comparative analysis of populations of samples. In practice, many factors resulting from the environment, the protocol, and even the instrument itself, can lead to minor discrepancies between spectra, rendering automated comparative analysis difficult. In this work, a sequence/pipeline of algorithms to correct variations between spectra is proposed. The algorithms correct multiple spectra by identifying peaks that are common to all and, from those, computes a spectrum-specific correction. We show that these algorithms increase comparability within large datasets of spectra, facilitating comparative analysis, such as machine learning.
Conflict of interest statement
The authors declare no competing interests.
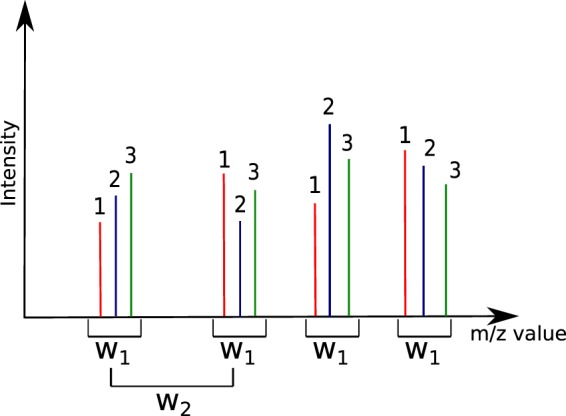
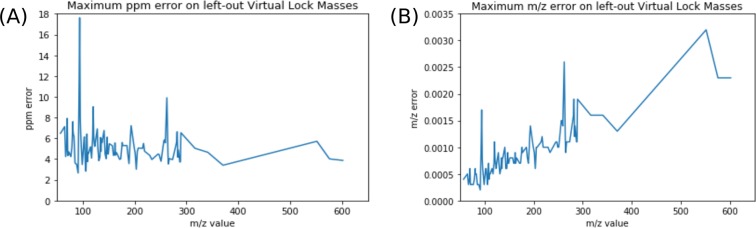
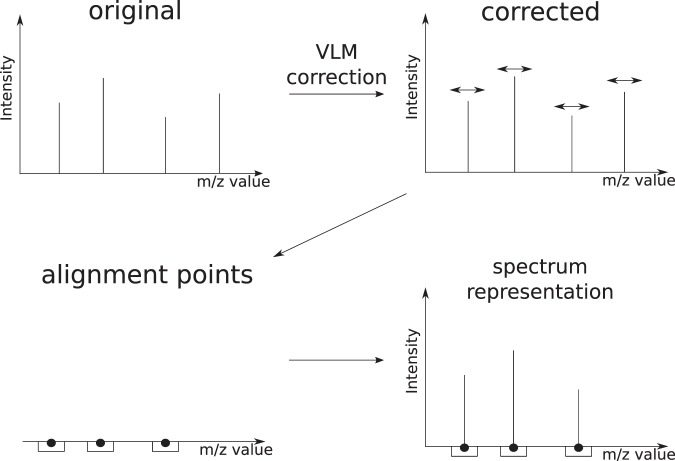
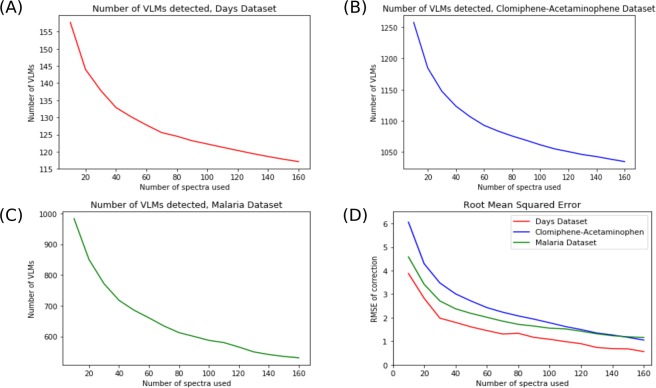
Figures


References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
