

DEGS1 variant causes neurological disorder
- PMID: 31186544
- PMCID: PMC6871177
- DOI: 10.1038/s41431-019-0444-z
DEGS1 variant causes neurological disorder
Abstract
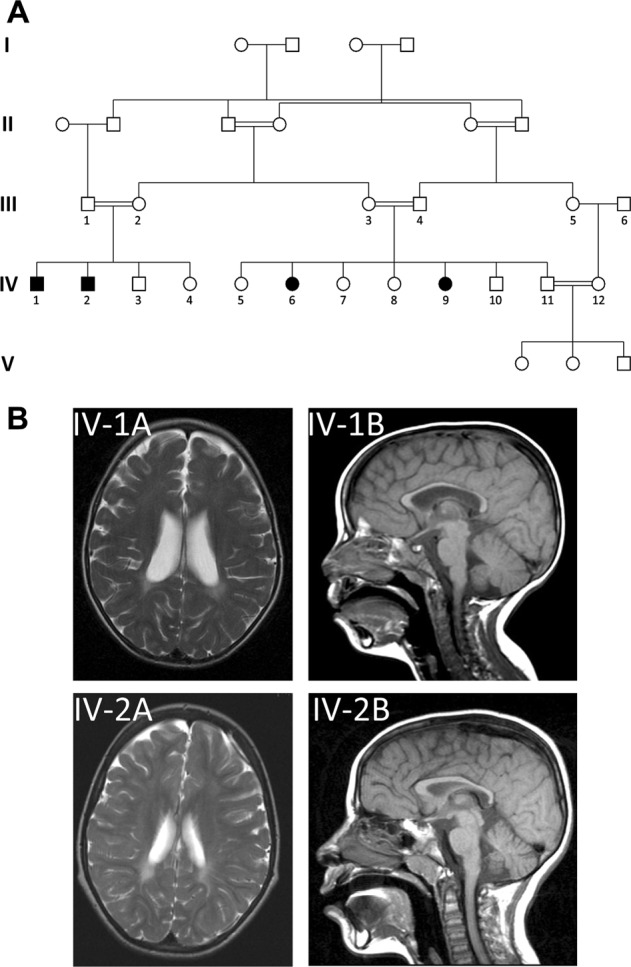
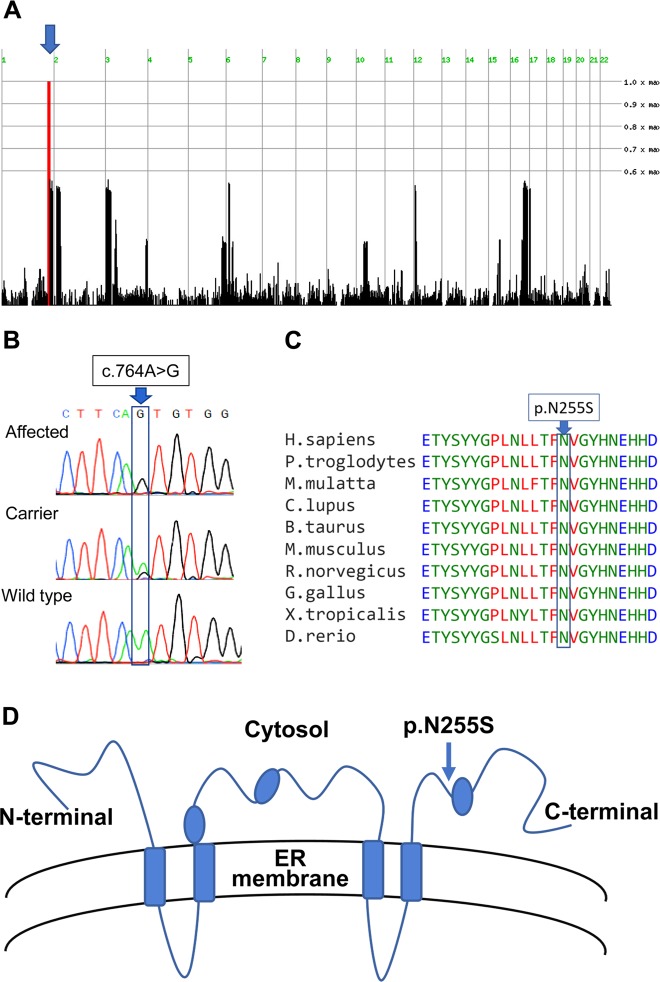
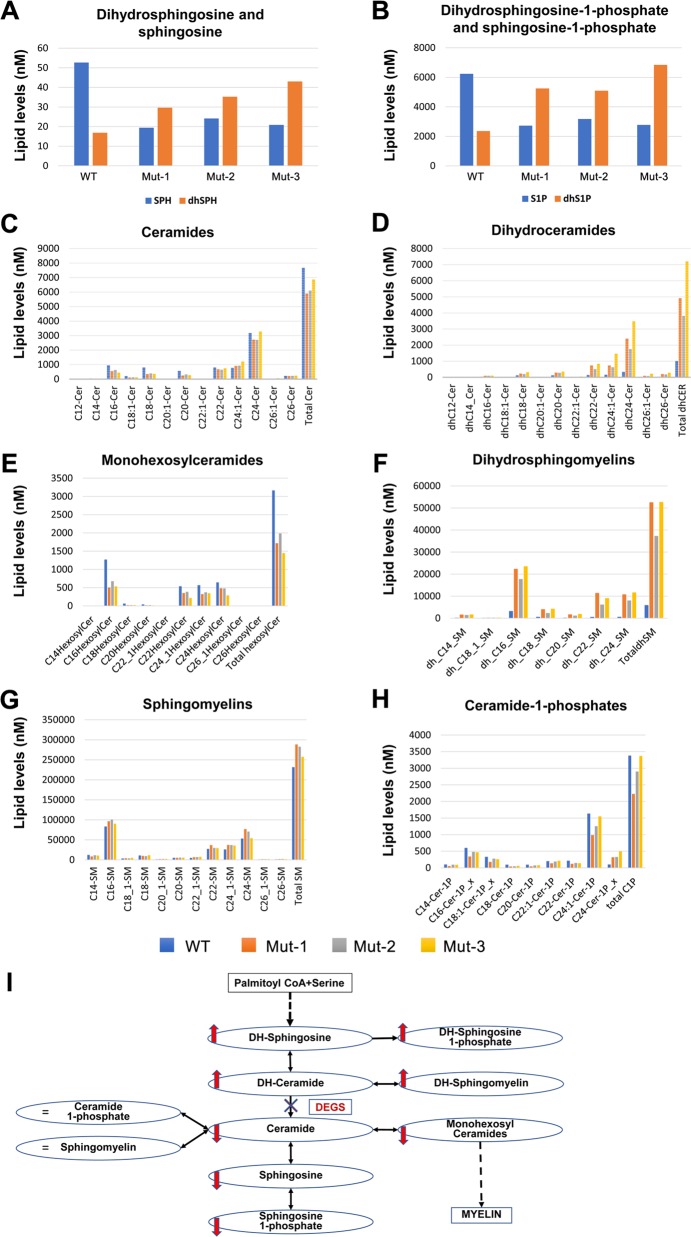
Sphingolipidoses are monogenic lipid storage diseases caused by variants in enzymes of lipid synthesis and metabolism. We describe an autosomal recessive complex neurological disorder affecting consanguineous kindred. All four affected individuals, born at term following normal pregnancies, had mild to severe intellectual disability, spastic quadriplegia, scoliosis and epilepsy in most, with no dysmorphic features. Brain MRI findings were suggestive of leukodystrophy, with abnormal hyperintense signal in the periventricular perioccipital region and thinning of the body of corpus callosum. Notably, all affected individuals were asymptomatic at early infancy and developed normally until the age of 8-18 months, when deterioration ensued. Homozygosity mapping identified a single 8.7 Mb disease-associated locus on chromosome 1q41-1q42.13 between rs1511695 and rs537250 (two-point LOD score 2.1). Whole exome sequencing, validated through Sanger sequencing, identified within this locus a single disease-associated homozygous variant in DEGS1, encoding C4-dihydroceramide desaturase, an enzyme of the ceramide synthesis pathway. The missense variant, segregating within the family as expected for recessive heredity, affects an evolutionary-conserved amino acid of all isoforms of DEGS1 (c.656A>G, c.764A>G; p.(N219S), p.(N255S)) and was not found in a homozygous state in ExAC and gnomAD databases or in 300 ethnically matched individuals. Lipidomcs analysis of whole blood of affected individuals demonstrated augmented levels of dihydroceramides, dihydrosphingosine, dihydrosphingosine-1-phosphate and dihydrosphingomyelins with reduced levels of ceramide, sphingosine, sphingosine-1-phosphate and monohexosylceramides, as expected in malfunction of C4-dihydroceramide desaturase. Thus, we describe a sphingolipidosis causing a severe regressive neurological disease.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
