

A pan-cancer analysis of synonymous mutations
- PMID: 31189880
- PMCID: PMC6562042
- DOI: 10.1038/s41467-019-10489-2
A pan-cancer analysis of synonymous mutations
Abstract
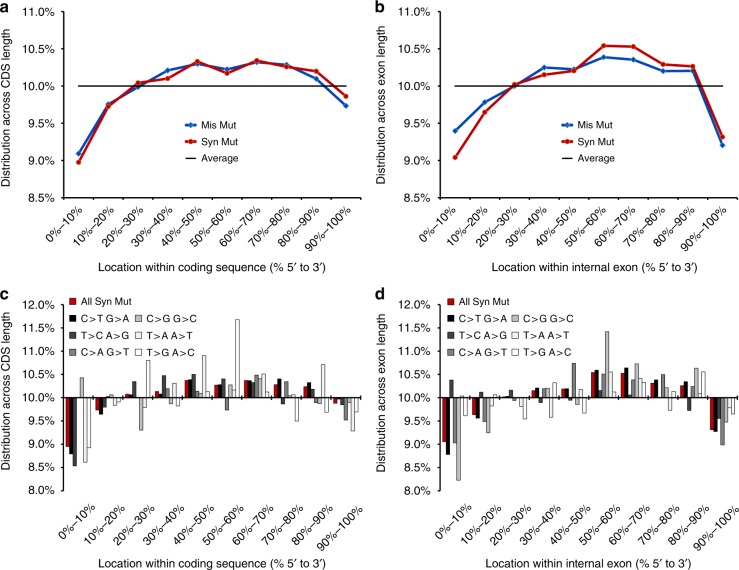
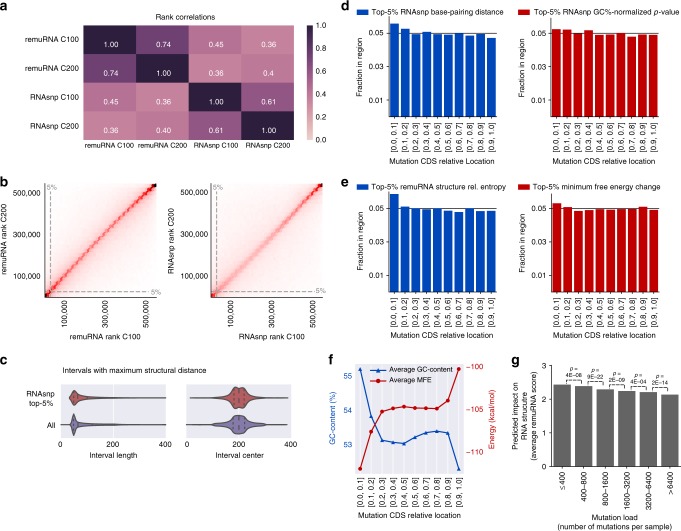
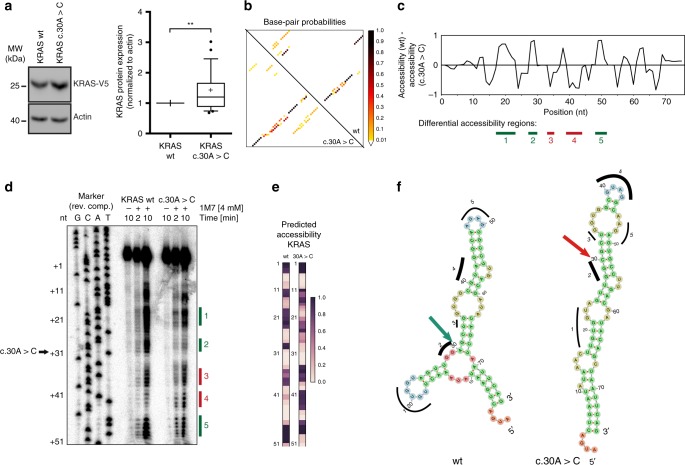
Synonymous mutations have been viewed as silent mutations, since they only affect the DNA and mRNA, but not the amino acid sequence of the resulting protein. Nonetheless, recent studies suggest their significant impact on splicing, RNA stability, RNA folding, translation or co-translational protein folding. Hence, we compile 659194 synonymous mutations found in human cancer and characterize their properties. We provide the user-friendly, comprehensive resource for synonymous mutations in cancer, SynMICdb ( http://SynMICdb.dkfz.de ), which also contains orthogonal information about gene annotation, recurrence, mutation loads, cancer association, conservation, alternative events, impact on mRNA structure and a SynMICdb score. Notably, synonymous and missense mutations are depleted at the 5'-end of the coding sequence as well as at the ends of internal exons independent of mutational signatures. For patient-derived synonymous mutations in the oncogene KRAS, we indicate that single point mutations can have a relevant impact on expression as well as on mRNA secondary structure.
Conflict of interest statement
S.D. is co-owner of the siTOOLs Biotech GmbH, Martinsried, Germany, without any relation to this study or competing financial interests. The remaining authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
