

A novel mutation of the PAX3 gene in a Chinese family with Waardenburg syndrome type I
- PMID: 31190477
- PMCID: PMC6625151
- DOI: 10.1002/mgg3.798
A novel mutation of the PAX3 gene in a Chinese family with Waardenburg syndrome type I
Abstract
Background: To analyze the clinical phenotypes and genetic variants of a Chinese family with Waardenburg syndrome (WS) and to explore the possible molecular pathogenesis of WS.
Methods: The clinical data from a patient and his family were collected. The genomic DNA of the patient and his family was purified from their peripheral blood. All exons and flanking sequences of the MITF, PAX3, SOX10, SNAI2, END3, and EDNRB genes were investigated through high-throughput sequencing. Based on the results of high-throughput sequencing, genetic variants in the patient and his family were verified and analyzed by Sanger sequencing.
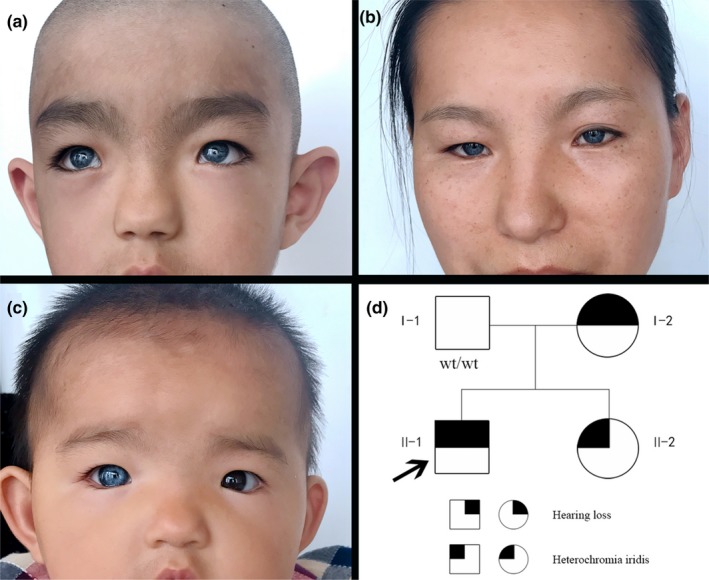
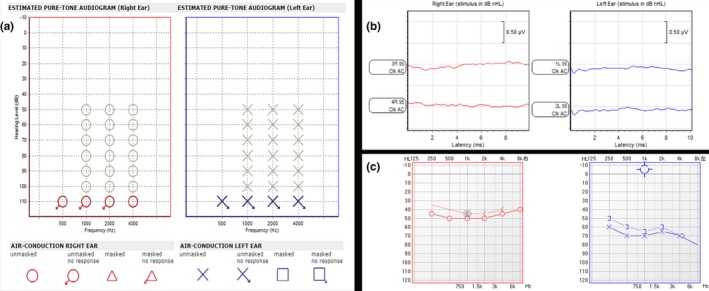
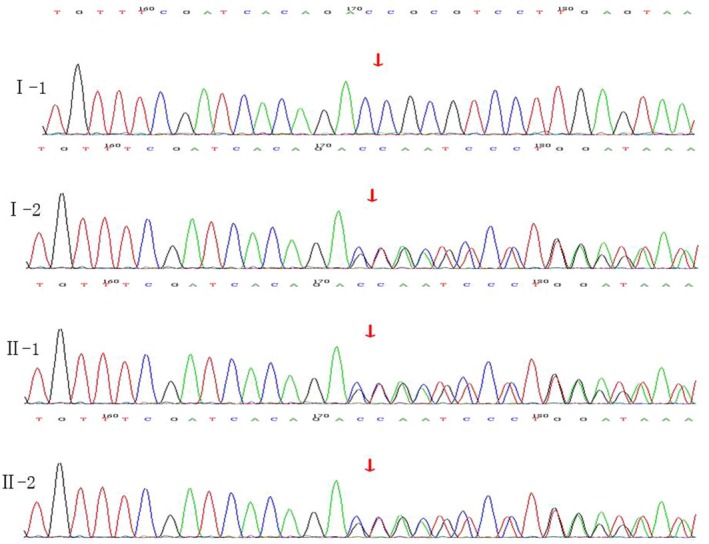
Results: The patient was diagnosed with typical WS1 that manifested in hearing impairment, inner canthus ectopia and heterochromic iris. Sanger sequencing revealed the pathogenic heterozygous c.420-424de1CGCGGinsTTAC mutation in the PAX3 gene in the proband, which is a frameshift mutation that changed the amino acid sequence of the PAX3 protein from AVCDRNTVPSV to YSVIETPCRQ* (* refers to a stop codon) from amino acids 141-151. The stop codon induced by this mutation resulted in the truncation of the PAX3 protein. The same mutation sites were also found in the mother and younger sister of the proband. No previous report of this mutation was found in the Human Gene Mutation Database.
Conclusion: The novel heterozygous c.420-424de1CGCGGinsTTAC mutation is the molecular pathological cause for WS1 in our patient. The clinical and genetic characterization of this family with WS1 elucidated the genetic heterogeneity of PAX3 in WS1. Moreover, the mutation detected in this case has expanded the database of PAX3 mutations.
Keywords: PAX3; Waardenburg syndrome type I; gene mutation; hereditary deafness.
© 2019 The Authors. Molecular Genetics & Genomic Medicine published by Wiley Periodicals, Inc.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Chen, H. , Jiang, L. U. , Xie, Z. , Mei, L. , He, C. , Hu, Z. , … Feng, Y. (2010). Novel mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or type II Waardenburg syndrome. Biochemical and Biophysical Research Communications, 397(1), 70–74. 10.1016/j.bbrc.2010.05.066 - DOI - PubMed
-
- de Sousa Andrade, S. M. , Monteiro, A. R. , Martins, J. H. , Alves, M. C. , Santos Silva, L. F. , Quadros, J. M. , & Ribeiro, C. A. (2012). Cochlear implant rehabilitation outcomes in Waardenburg syndrome children. International Journal of Pediatric Otorhinolaryngology, 76(9), 1375–1378. 10.1016/j.ijporl.2012.06.010 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
