

Epidermolytic hyperkeratosis: clinical update
- PMID: 31190940
- PMCID: PMC6512611
- DOI: 10.2147/CCID.S166849
Epidermolytic hyperkeratosis: clinical update
Abstract
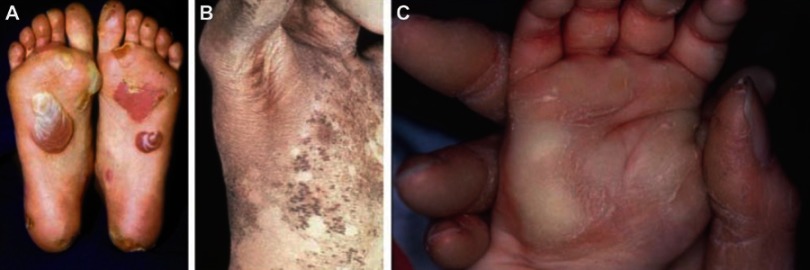
Epidermolytic hyperkeratosis (EHK), earlier termed as bullous congenital ichthyosiform erythroderma is a skin disorder characterized as an autosomal dominant and rare disorder which has been observed to affect 1 in over 200,000 infants as a consequence of a significant mutation in the genes responsible for the keratin proteins, mostly keratin 1 and 10. The features present at birth include erythema and blistering. In adults, the hallmarks include hyperkeratosis, erosions, and blisters. The major symptoms including xerosis, pruritus, and painful fissuring lead not only to cosmetic problems but also stress, inferiority complex and other psychological conditions. While clinical inspection followed by confirmatory tests including histopathology and electron microscopic assessment is used for diagnosis, treatment modalities can be further improved for better diagnosis. This article reviews subtypes of ichthyosis, with a focus on EHK, genetics behind the disease, recently reported mutations, the existing diagnostics and treatments for the same and potential of new modalities in diagnosis/treatment.
Keywords: epidermolytic hyperkeratosis; ichthyosis; skin; skin disorder.
Conflict of interest statement
The authors report no conflicts of interest in this work.
Figures
References
-
- Goldsmith LAJPMG. The ichthyosis. Prog Med Genet. 1976;1:185–240. - PubMed
-
- Esterly NBJP. The ichthyosiform dermatoses. Pediatrics. 1968;42(6):990–1004. - PubMed
-
- Hirone TJM. Electron microscopic studies of ichthyosis and congenital ichthyosiform erythroderma. J Electron Microsc (Tokyo). 1969;18(1):63–72. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
