

Loss-of-function mutations in Lysyl-tRNA synthetase cause various leukoencephalopathy phenotypes
- PMID: 31192300
- PMCID: PMC6515944
- DOI: 10.1212/NXG.0000000000000316
Loss-of-function mutations in Lysyl-tRNA synthetase cause various leukoencephalopathy phenotypes
Abstract
Objective: To expand the clinical spectrum of lysyl-tRNA synthetase (KARS) gene-related diseases, which so far includes Charcot-Marie-Tooth disease, congenital visual impairment and microcephaly, and nonsyndromic hearing impairment.
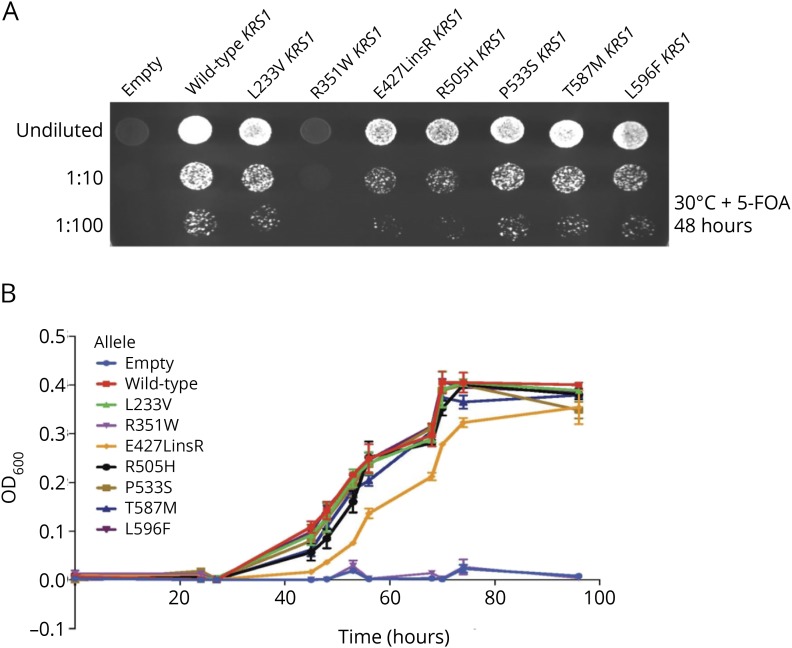
Methods: Whole-exome sequencing was performed on index patients from 4 unrelated families with leukoencephalopathy. Candidate pathogenic variants and their cosegregation were confirmed by Sanger sequencing. Effects of mutations on KARS protein function were examined by aminoacylation assays and yeast complementation assays.
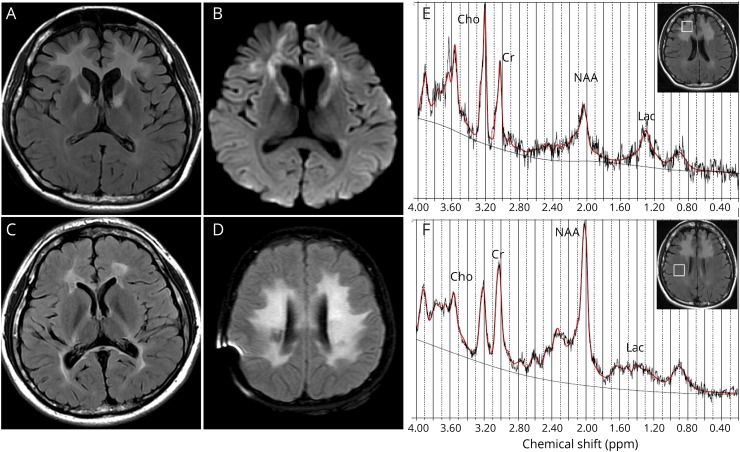
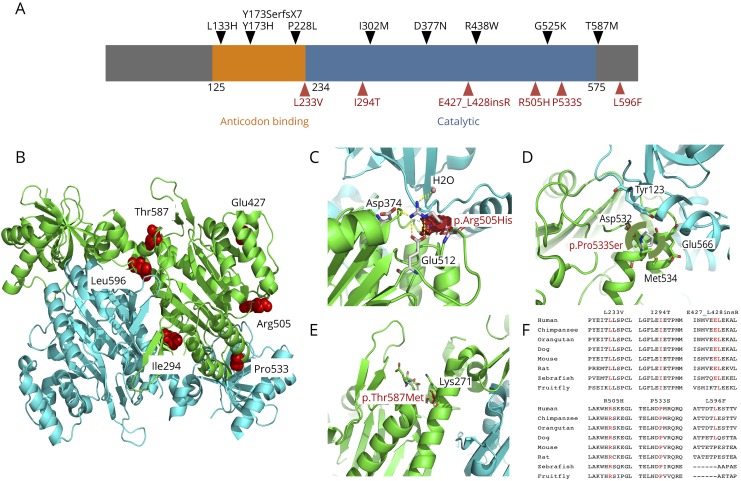
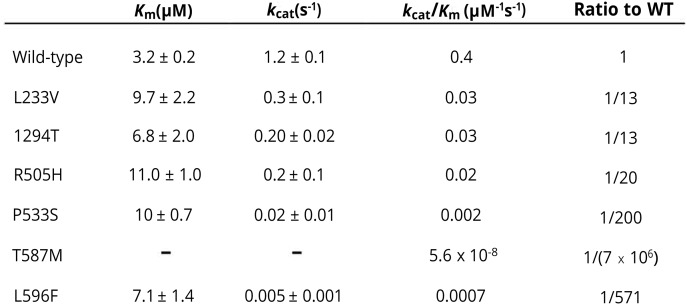
Results: Common clinical features of the patients in this study included impaired cognitive ability, seizure, hypotonia, ataxia, and abnormal brain imaging, suggesting that the CNS involvement is the main clinical presentation. Six previously unreported and 1 known KARS mutations were identified and cosegregated in these families. Two patients are compound heterozygous for missense mutations, 1 patient is homozygous for a missense mutation, and 1 patient harbored an insertion mutation and a missense mutation. Functional and structural analyses revealed that these mutations impair aminoacylation activity of lysyl-tRNA synthetase, indicating that defective KARS function is responsible for the phenotypes in these individuals.
Conclusions: Our results demonstrate that patients with loss-of-function KARS mutations can manifest CNS disorders, thus broadening the phenotypic spectrum associated with KARS-related disease.
Figures
Similar articles
-
Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89.Am J Hum Genet. 2013 Jul 11;93(1):132-40. doi: 10.1016/j.ajhg.2013.05.018. Epub 2013 Jun 13. Am J Hum Genet. 2013. PMID: 23768514 Free PMC article.
-
Biallelic KARS pathogenic variants cause an early-onset progressive leukodystrophy.Brain. 2019 Mar 1;142(3):560-573. doi: 10.1093/brain/awz001. Brain. 2019. PMID: 30715177
-
Mutations in KARS cause early-onset hearing loss and leukoencephalopathy: Potential pathogenic mechanism.Hum Mutat. 2017 Dec;38(12):1740-1750. doi: 10.1002/humu.23335. Epub 2017 Sep 29. Hum Mutat. 2017. PMID: 28887846
-
KARS-related diseases: progressive leukoencephalopathy with brainstem and spinal cord calcifications as new phenotype and a review of literature.Orphanet J Rare Dis. 2018 Apr 4;13(1):45. doi: 10.1186/s13023-018-0788-4. Orphanet J Rare Dis. 2018. PMID: 29615062 Free PMC article. Review.
-
A novel compound heterozygous missense mutation in ASNS broadens the spectrum of asparagine synthetase deficiency.Mol Genet Genomic Med. 2020 Jun;8(6):e1235. doi: 10.1002/mgg3.1235. Epub 2020 Apr 7. Mol Genet Genomic Med. 2020. PMID: 32255274 Free PMC article. Review.
Cited by
-
Mitochondrial Aminoacyl-tRNA Synthetase and Disease: The Yeast Contribution for Functional Analysis of Novel Variants.Int J Mol Sci. 2021 Apr 26;22(9):4524. doi: 10.3390/ijms22094524. Int J Mol Sci. 2021. PMID: 33926074 Free PMC article. Review.
-
Recessive aminoacyl-tRNA synthetase disorders: lessons learned from in vivo disease models.Front Neurosci. 2023 May 9;17:1182874. doi: 10.3389/fnins.2023.1182874. eCollection 2023. Front Neurosci. 2023. PMID: 37274208 Free PMC article. Review.
-
Novel Cases of Non-Syndromic Hearing Impairment Caused by Pathogenic Variants in Genes Encoding Mitochondrial Aminoacyl-tRNA Synthetases.Genes (Basel). 2024 Jul 19;15(7):951. doi: 10.3390/genes15070951. Genes (Basel). 2024. PMID: 39062730 Free PMC article.
-
A Cysteinyl-tRNA Synthetase Mutation Causes Novel Autosomal-Dominant Inheritance of a Parkinsonism/Spinocerebellar-Ataxia Complex.Neurosci Bull. 2024 Oct;40(10):1489-1501. doi: 10.1007/s12264-024-01231-0. Epub 2024 Jun 13. Neurosci Bull. 2024. PMID: 38869703 Free PMC article.
-
Untargeted metabolomics to evaluate polymyxin B toxicodynamics following direct intracerebroventricular administration into the rat brain.Comput Struct Biotechnol J. 2022 Nov 7;20:6067-6077. doi: 10.1016/j.csbj.2022.10.041. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36420146 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous