

Delivering on the promise of gene editing for cystic fibrosis
- PMID: 31193992
- PMCID: PMC6545485
- DOI: 10.1016/j.gendis.2018.11.005
Delivering on the promise of gene editing for cystic fibrosis
Abstract
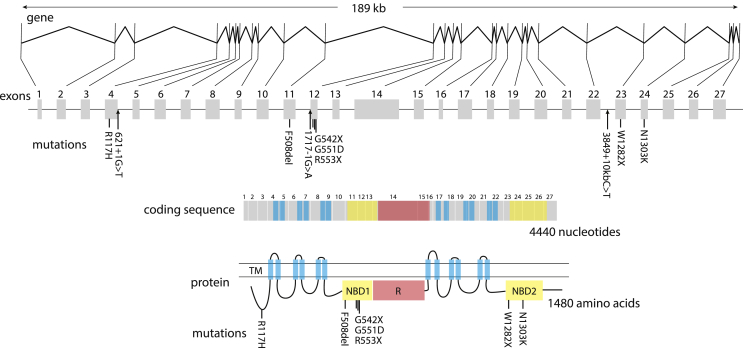
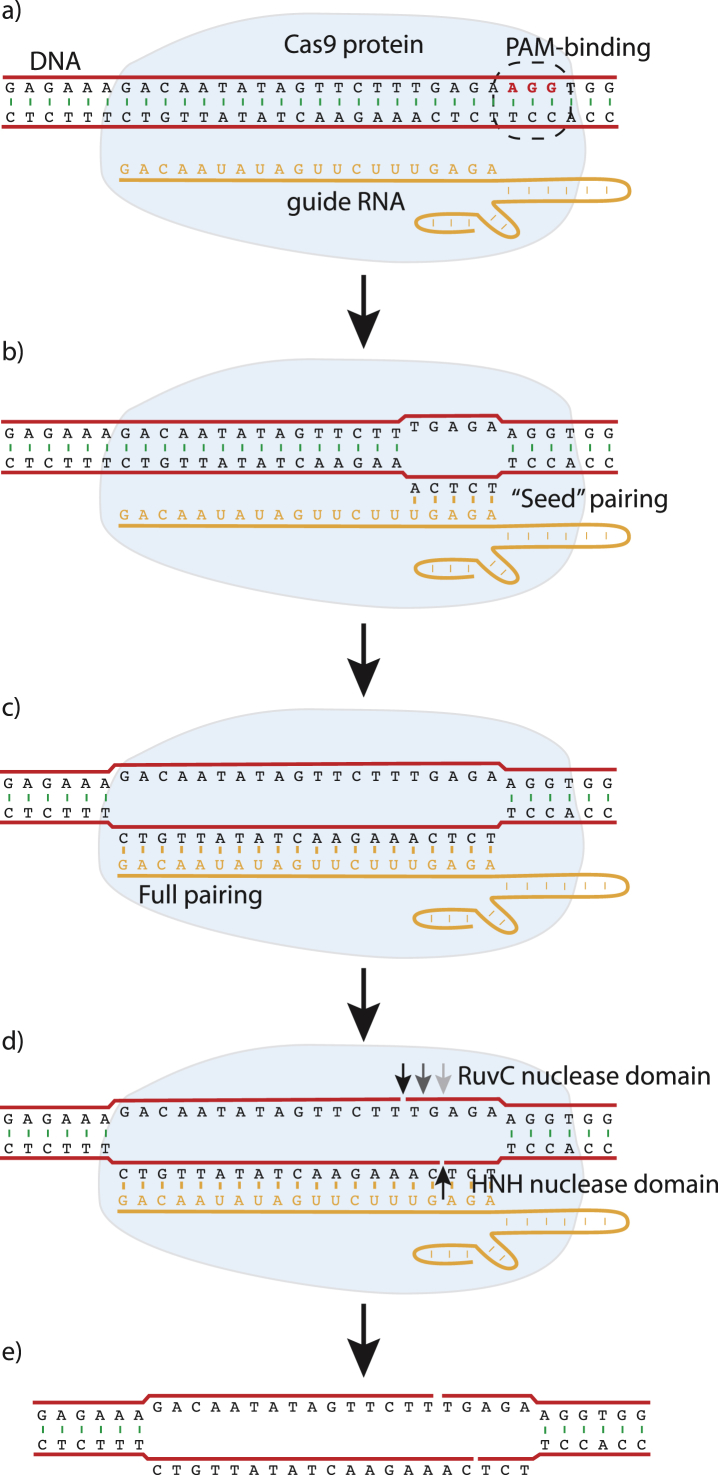
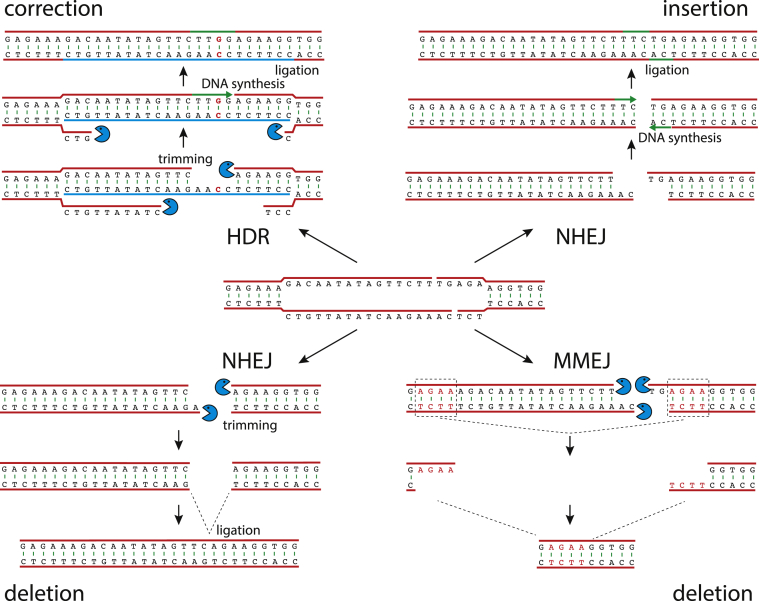
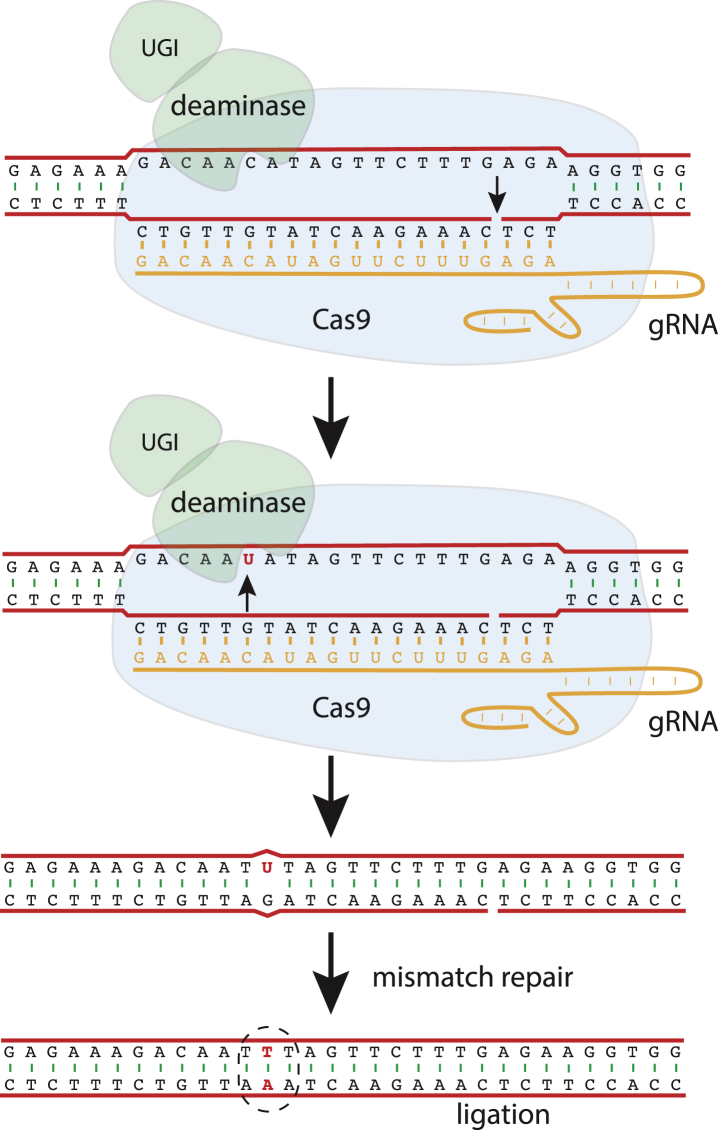
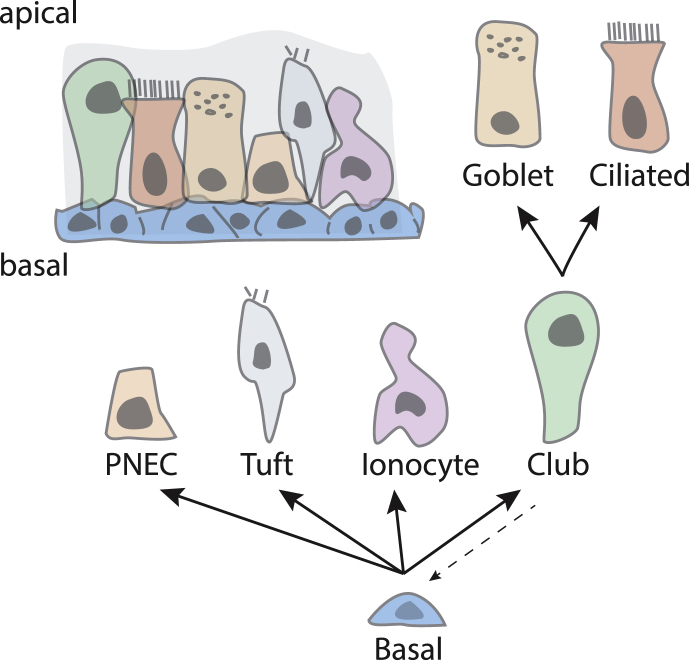
In this review, we describe a path for translation of gene editing into therapy for cystic fibrosis (CF). Cystic fibrosis results from mutations in the CFTR gene, with one allele predominant in patient populations. This simple, genetic etiology makes gene editing appealing for treatment of this disease. There already have been success in applying this approach to cystic fibrosis in cell and animal models, although these advances have been modest in comparison to advances for other disease. Less than six years after its first demonstration in animals, CRISPR/Cas gene editing is in early clinical trials for several disorders. Most clinical trials, thus far, attempt to edit genes in cells of the blood lineages. The advantage of the blood is that the stem cells are known, can be isolated, edited, selected, expanded, and returned to the body. The likely next trials will be in the liver, which is accessible to many delivery methods. For cystic fibrosis, the biggest hurdle is to deliver editors to other, less accessible organs. We outline a path by which delivery can be improved. The translation of new therapies doesn't occur in isolation, and the development of gene editors is occurring as advances in gene therapy and small molecule therapeutics are being made. The advances made in gene therapy may help develop delivery vehicles for gene editing, although major improvements are needed. Conversely, the approval of effective small molecule therapies for many patients with cystic fibrosis will raise the bar for translation of gene editing.
Keywords: CFTR gene; CRISPR/Cas9; Cystic fibrosis; Gene editing; Gene therapy.
Figures
Similar articles
-
In Vitro Validation of a CRISPR-Mediated CFTR Correction Strategy for Preclinical Translation in Pigs.Hum Gene Ther. 2019 Sep;30(9):1101-1116. doi: 10.1089/hum.2019.074. Epub 2019 Jun 18. Hum Gene Ther. 2019. PMID: 31099266
-
Potential of helper-dependent Adenoviral vectors in CRISPR-cas9-mediated lung gene therapy.Cell Biosci. 2021 Jul 23;11(1):145. doi: 10.1186/s13578-021-00662-w. Cell Biosci. 2021. PMID: 34301308 Free PMC article. Review.
-
A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene.JCI Insight. 2018 Oct 4;3(19):e123529. doi: 10.1172/jci.insight.123529. JCI Insight. 2018. PMID: 30282831 Free PMC article.
-
Innovative Therapeutic Strategies for Cystic Fibrosis: Moving Forward to CRISPR Technique.Front Pharmacol. 2018 Apr 20;9:396. doi: 10.3389/fphar.2018.00396. eCollection 2018. Front Pharmacol. 2018. PMID: 29731717 Free PMC article. Review.
-
Transcriptome Profiling and Molecular Therapeutic Advances in Cystic Fibrosis: Recent Insights.Genes (Basel). 2019 Feb 26;10(3):180. doi: 10.3390/genes10030180. Genes (Basel). 2019. PMID: 30813620 Free PMC article. Review.
Cited by
-
Exosome-mediated delivery of Cas9 ribonucleoprotein complexes for tissue-specific gene therapy of liver diseases.Sci Adv. 2022 Sep 16;8(37):eabp9435. doi: 10.1126/sciadv.abp9435. Epub 2022 Sep 14. Sci Adv. 2022. PMID: 36103526 Free PMC article.
-
Acidic Submucosal Gland pH and Elevated Protein Concentration Produce Abnormal Cystic Fibrosis Mucus.Dev Cell. 2020 Aug 24;54(4):488-500.e5. doi: 10.1016/j.devcel.2020.07.002. Epub 2020 Jul 29. Dev Cell. 2020. PMID: 32730755 Free PMC article.
-
Adeno-Associated Virus Mediated Gene Therapy for Corneal Diseases.Pharmaceutics. 2020 Aug 13;12(8):767. doi: 10.3390/pharmaceutics12080767. Pharmaceutics. 2020. PMID: 32823625 Free PMC article. Review.
-
Anticipating New Treatments for Cystic Fibrosis: A Global Survey of Researchers.J Clin Med. 2022 Feb 26;11(5):1283. doi: 10.3390/jcm11051283. J Clin Med. 2022. PMID: 35268374 Free PMC article.
-
CRISPR, Prime Editing, Optogenetics, and DREADDs: New Therapeutic Approaches Provided by Emerging Technologies in the Treatment of Spinal Cord Injury.Mol Neurobiol. 2020 Apr;57(4):2085-2100. doi: 10.1007/s12035-019-01861-w. Epub 2020 Jan 11. Mol Neurobiol. 2020. PMID: 31927725 Review.
References
-
- Bobadilla J.L., Macek M., Jr., Fine J.P., Farrell P.M. Cystic fibrosis: a worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606. - PubMed
-
- Egan M.E. Genetics of cystic fibrosis: clinical implications. Clin Chest Med. 2016;37(1):9–16. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
