

Metabolic network percolation quantifies biosynthetic capabilities across the human oral microbiome
- PMID: 31194675
- PMCID: PMC6609349
- DOI: 10.7554/eLife.39733
Metabolic network percolation quantifies biosynthetic capabilities across the human oral microbiome
Abstract
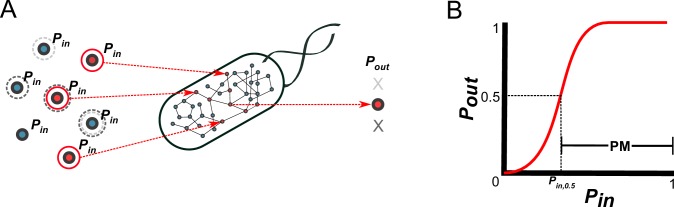
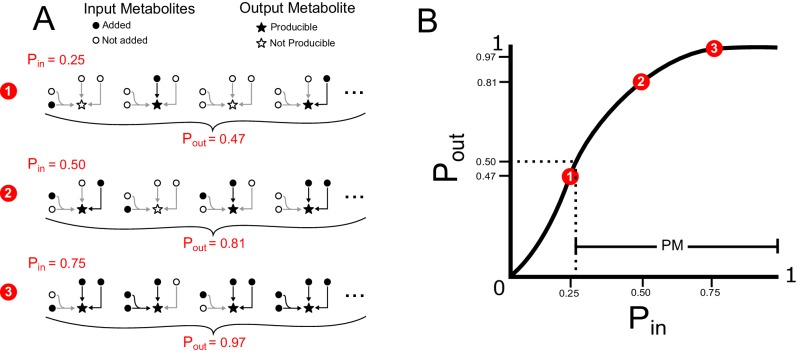
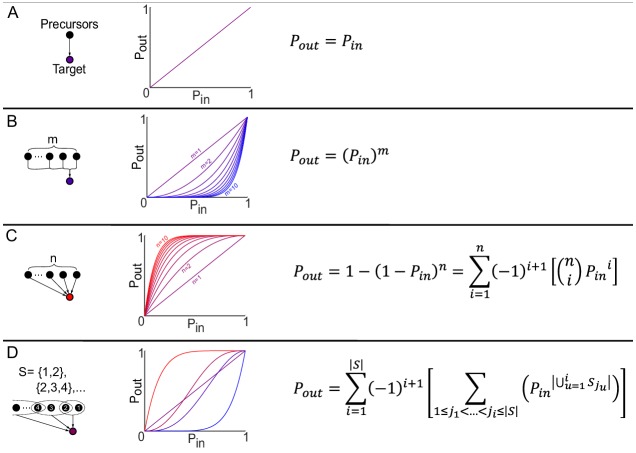
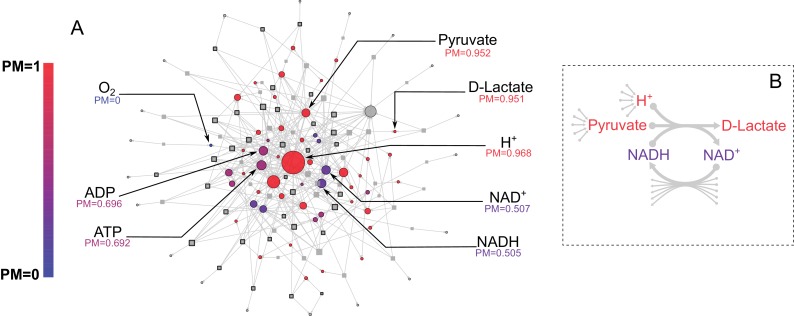
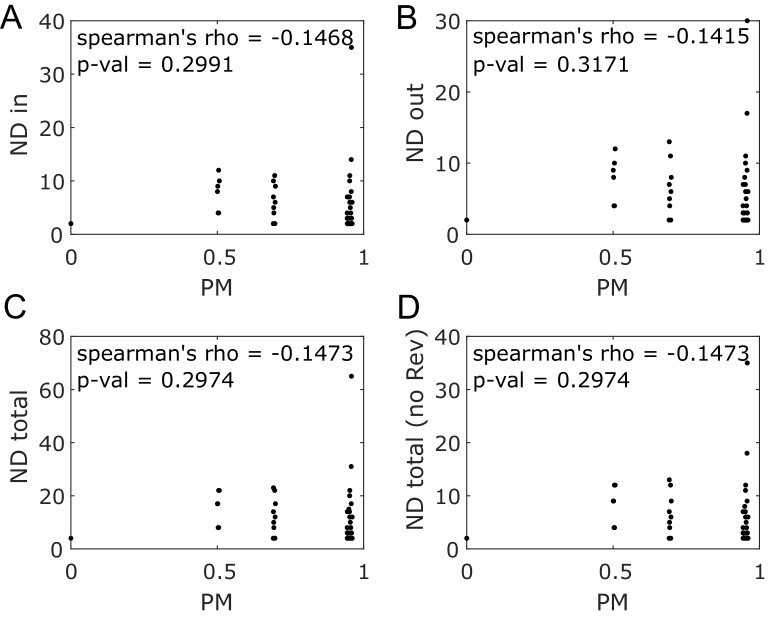
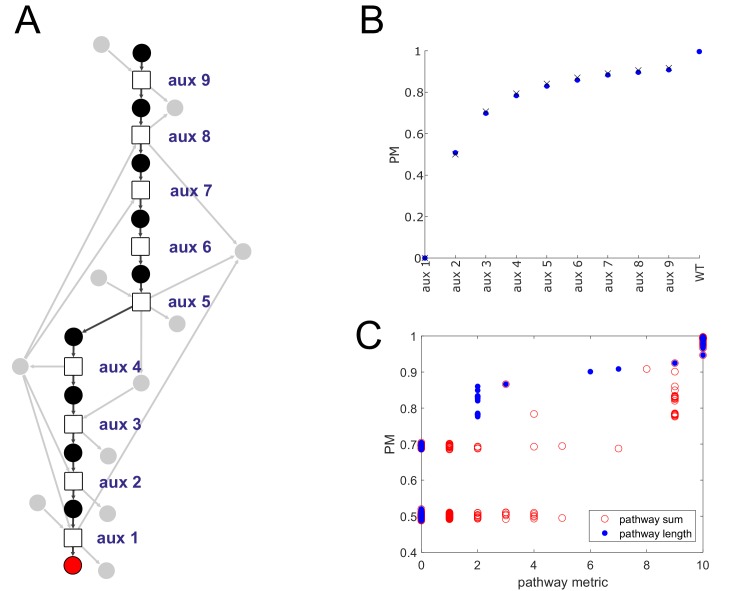
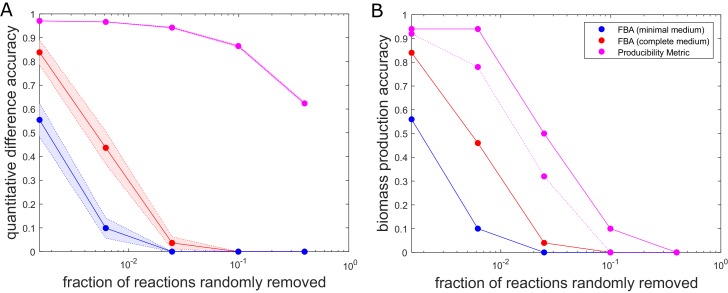
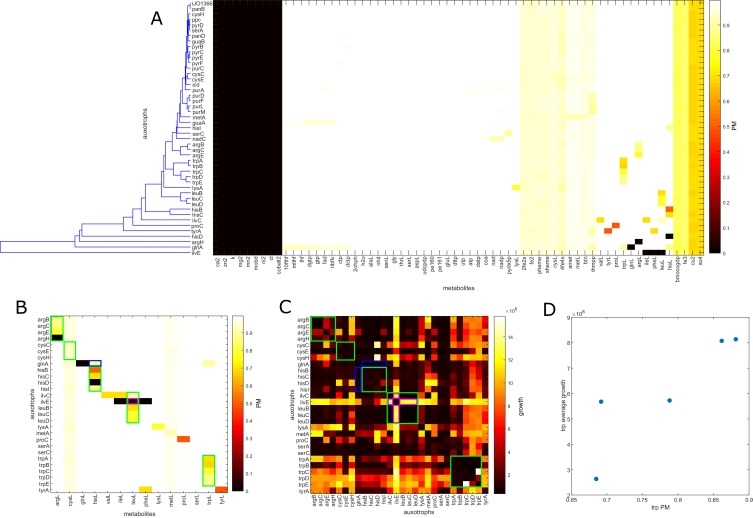
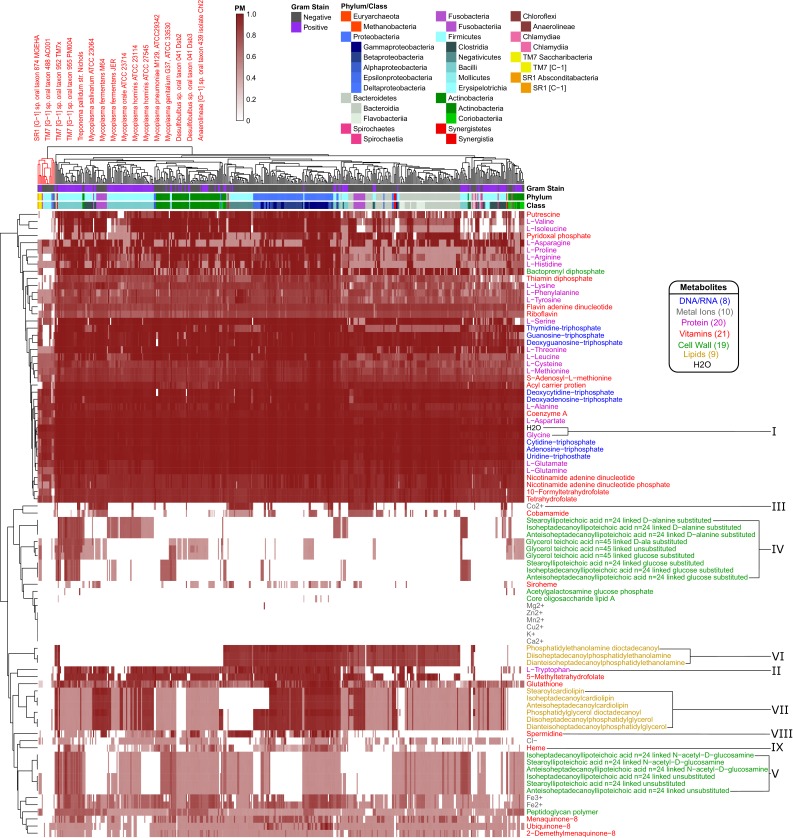
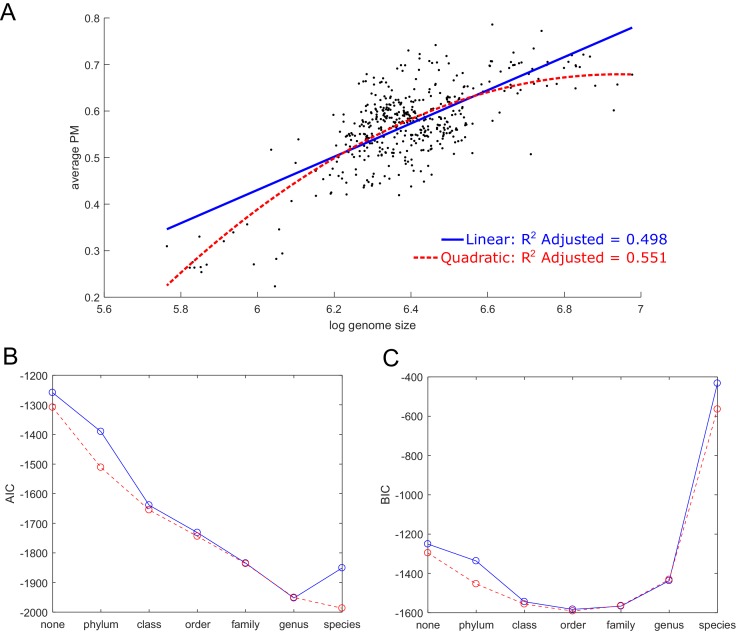
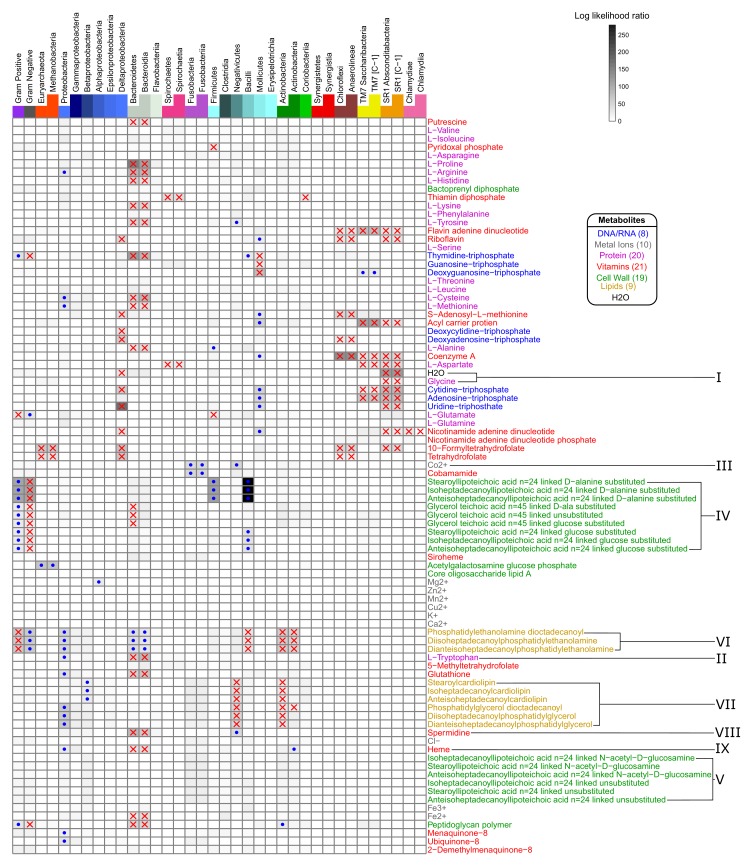
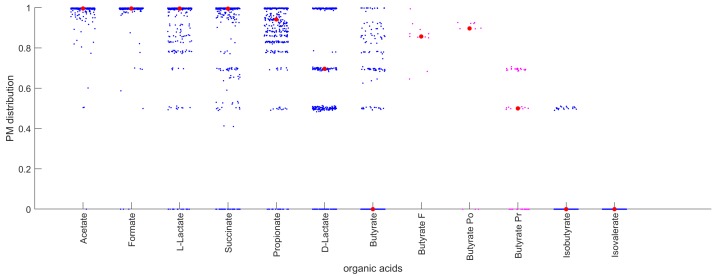
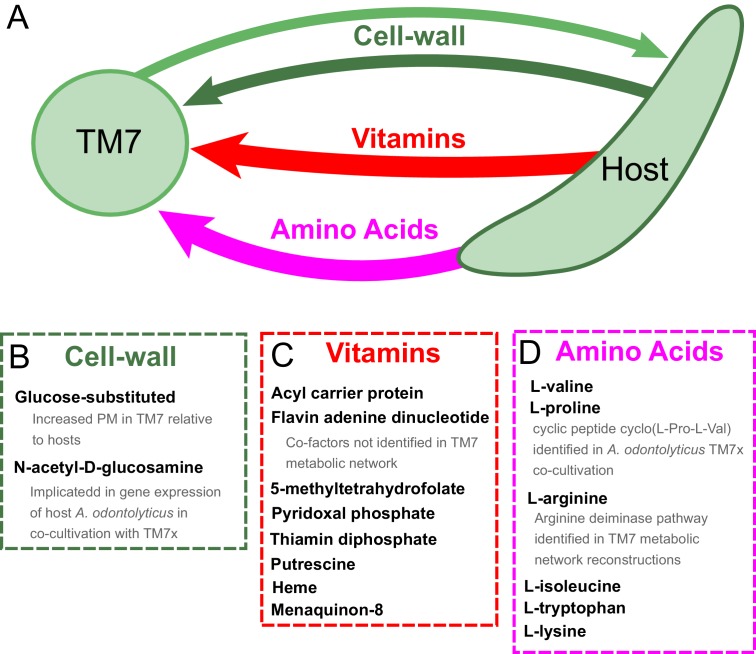
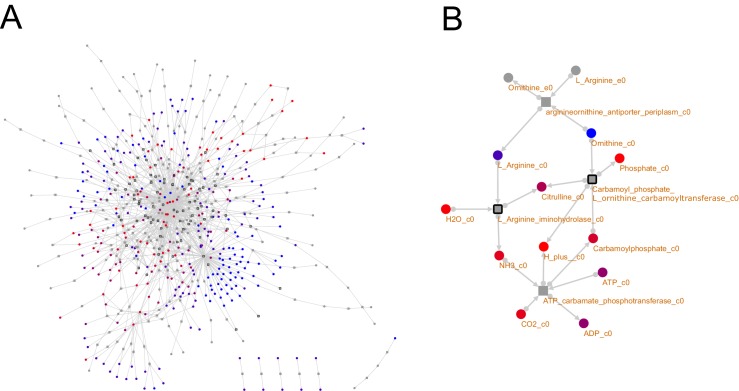
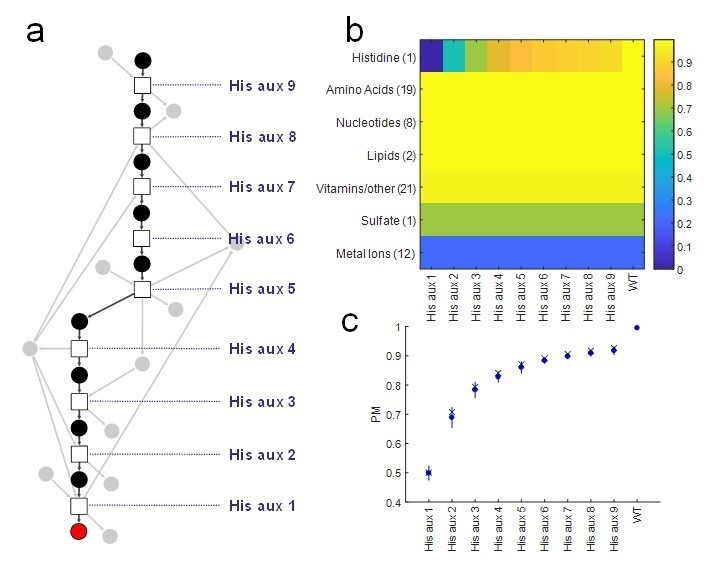
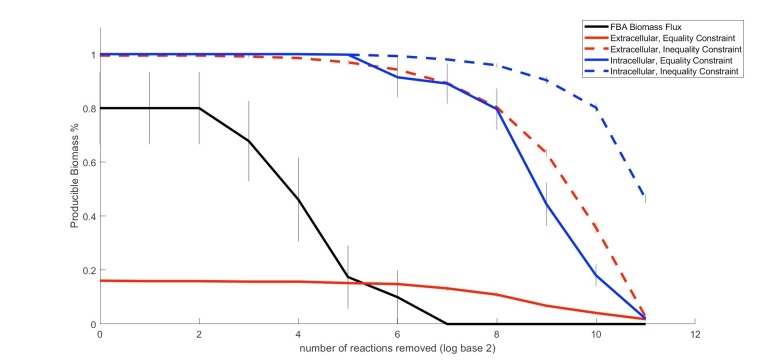
The biosynthetic capabilities of microbes underlie their growth and interactions, playing a prominent role in microbial community structure. For large, diverse microbial communities, prediction of these capabilities is limited by uncertainty about metabolic functions and environmental conditions. To address this challenge, we propose a probabilistic method, inspired by percolation theory, to computationally quantify how robustly a genome-derived metabolic network produces a given set of metabolites under an ensemble of variable environments. We used this method to compile an atlas of predicted biosynthetic capabilities for 97 metabolites across 456 human oral microbes. This atlas captures taxonomically-related trends in biomass composition, and makes it possible to estimate inter-microbial metabolic distances that correlate with microbial co-occurrences. We also found a distinct cluster of fastidious/uncultivated taxa, including several Saccharibacteria (TM7) species, characterized by their abundant metabolic deficiencies. By embracing uncertainty, our approach can be broadly applied to understanding metabolic interactions in complex microbial ecosystems.
Keywords: Saccharibacteria (TM7); computational biology; human microbiome; metabolic modeling; oral microbial flora; oral prokaryotes; systems biology; uncultivated bacteria.
© 2019, Bernstein et al.
Conflict of interest statement
DB, FD, DS No competing interests declared
Figures

Similar articles
-
Functional and genetic markers of niche partitioning among enigmatic members of the human oral microbiome.Genome Biol. 2020 Dec 16;21(1):292. doi: 10.1186/s13059-020-02195-w. Genome Biol. 2020. PMID: 33323122 Free PMC article.
-
Advancements toward a systems level understanding of the human oral microbiome.Front Cell Infect Microbiol. 2014 Jul 29;4:98. doi: 10.3389/fcimb.2014.00098. eCollection 2014. Front Cell Infect Microbiol. 2014. PMID: 25120956 Free PMC article. Review.
-
Supragingival Plaque Microbiome Ecology and Functional Potential in the Context of Health and Disease.mBio. 2018 Nov 27;9(6):e01631-18. doi: 10.1128/mBio.01631-18. mBio. 2018. PMID: 30482830 Free PMC article.
-
Modeling metabolism of the human gut microbiome.Curr Opin Biotechnol. 2018 Jun;51:90-96. doi: 10.1016/j.copbio.2017.12.005. Epub 2017 Dec 16. Curr Opin Biotechnol. 2018. PMID: 29258014 Review.
-
Bacterial Community Assembly, Succession, and Metabolic Function during Outdoor Cultivation of Microchloropsis salina.mSphere. 2022 Aug 31;7(4):e0023122. doi: 10.1128/msphere.00231-22. Epub 2022 Jun 22. mSphere. 2022. PMID: 35730934 Free PMC article.
Cited by
-
Microbiome modeling: a beginner's guide.Front Microbiol. 2024 Jun 19;15:1368377. doi: 10.3389/fmicb.2024.1368377. eCollection 2024. Front Microbiol. 2024. PMID: 38962127 Free PMC article. Review.
-
Multi-genome metabolic modeling predicts functional inter-dependencies in the Arabidopsis root microbiome.Microbiome. 2022 Dec 9;10(1):217. doi: 10.1186/s40168-022-01383-z. Microbiome. 2022. PMID: 36482420 Free PMC article.
-
The oralome and its dysbiosis: New insights into oral microbiome-host interactions.Comput Struct Biotechnol J. 2021 Feb 27;19:1335-1360. doi: 10.1016/j.csbj.2021.02.010. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 33777334 Free PMC article. Review.
-
Seed2LP: seed inference in metabolic networks for reverse ecology applications.Bioinformatics. 2025 Mar 29;41(4):btaf140. doi: 10.1093/bioinformatics/btaf140. Bioinformatics. 2025. PMID: 40163742 Free PMC article.
-
Functional comparison of metabolic networks across species.Nat Commun. 2023 Mar 27;14(1):1699. doi: 10.1038/s41467-023-37429-5. Nat Commun. 2023. PMID: 36973280 Free PMC article.
References
-
- Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, Dehal P, Ware D, Perez F, Canon S, Sneddon MW, Henderson ML, Riehl WJ, Murphy-Olson D, Chan SY, Kamimura RT, Kumari S, Drake MM, Brettin TS, Glass EM, Chivian D, Gunter D, Weston DJ, Allen BH, Baumohl J, Best AA, Bowen B, Brenner SE, Bun CC, Chandonia JM, Chia JM, Colasanti R, Conrad N, Davis JJ, Davison BH, DeJongh M, Devoid S, Dietrich E, Dubchak I, Edirisinghe JN, Fang G, Faria JP, Frybarger PM, Gerlach W, Gerstein M, Greiner A, Gurtowski J, Haun HL, He F, Jain R, Joachimiak MP, Keegan KP, Kondo S, Kumar V, Land ML, Meyer F, Mills M, Novichkov PS, Oh T, Olsen GJ, Olson R, Parrello B, Pasternak S, Pearson E, Poon SS, Price GA, Ramakrishnan S, Ranjan P, Ronald PC, Schatz MC, Seaver SMD, Shukla M, Sutormin RA, Syed MH, Thomason J, Tintle NL, Wang D, Xia F, Yoo H, Yoo S, Yu D. KBase: the united states department of energy systems biology knowledgebase. Nature Biotechnology. 2018;36:566–569. doi: 10.1038/nbt.4163. - DOI - PMC - PubMed
-
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- T32GM008764/GM/NIGMS NIH HHS/United States
- T32 GM008764/GM/NIGMS NIH HHS/United States
- R01 DE024468/DE/NIDCR NIH HHS/United States
- R01 GM121950/GM/NIGMS NIH HHS/United States
- DE-SC0012627/Biological and Environmental Research/International
- RGP0020/2016/Human Frontier Science Program/International
- NSFOCE-BSF 1635070/National Science Foundation/International
- HR0011-15-C-0091/Defense Advanced Research Projects Agency/International
- R37DE016937/DE/NIDCR NIH HHS/United States
- R37 DE016937/DE/NIDCR NIH HHS/United States
- R01GM121950/GM/NIGMS NIH HHS/United States
- R01DE024468/DE/NIDCR NIH HHS/United States
- 1457695/National Science Foundation/International
LinkOut - more resources
Full Text Sources
