

Disentangling juxtacrine from paracrine signalling in dynamic tissue
- PMID: 31194728
- PMCID: PMC6592563
- DOI: 10.1371/journal.pcbi.1007030
Disentangling juxtacrine from paracrine signalling in dynamic tissue
Abstract
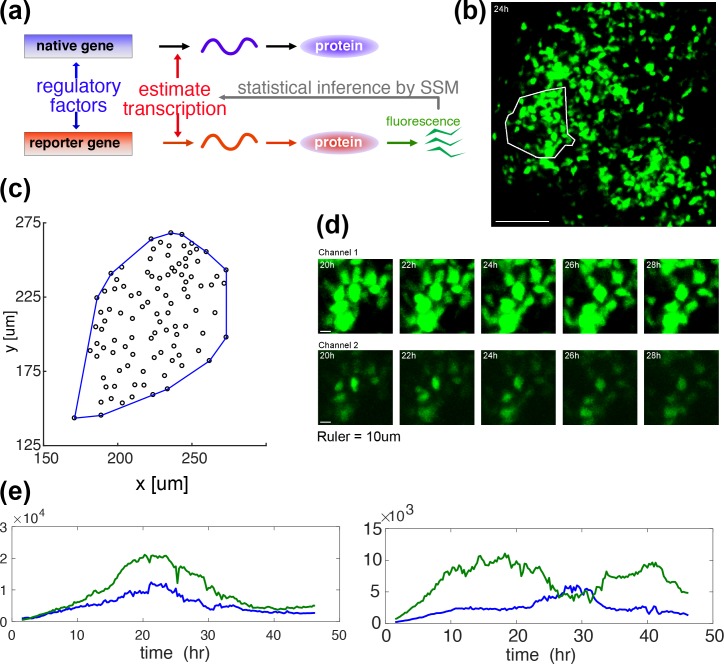
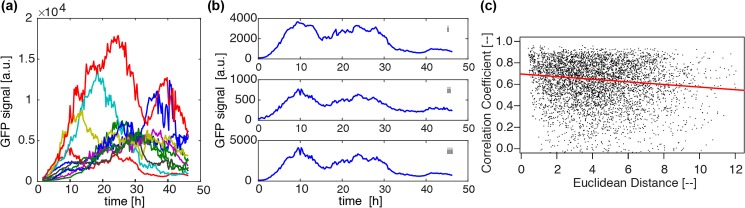
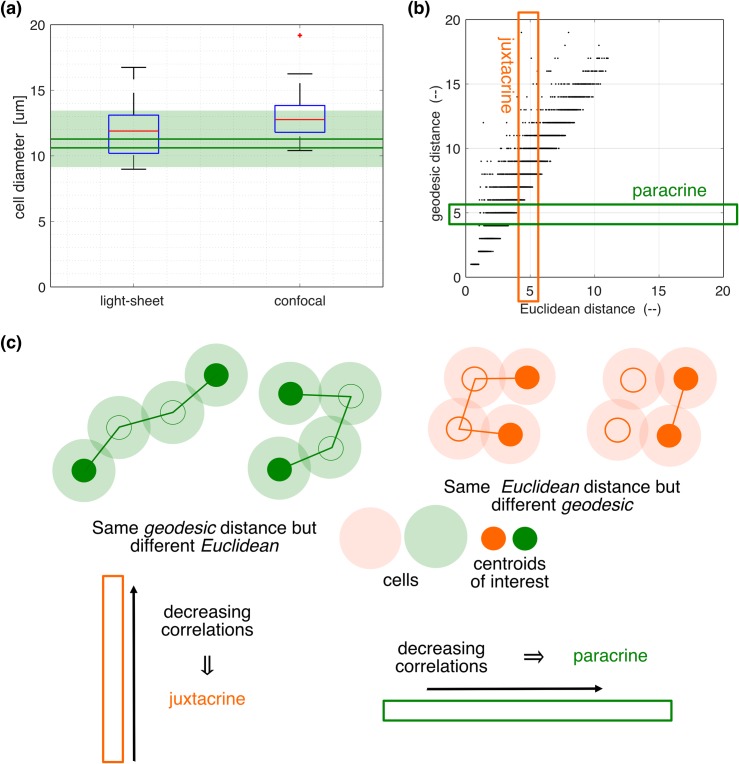
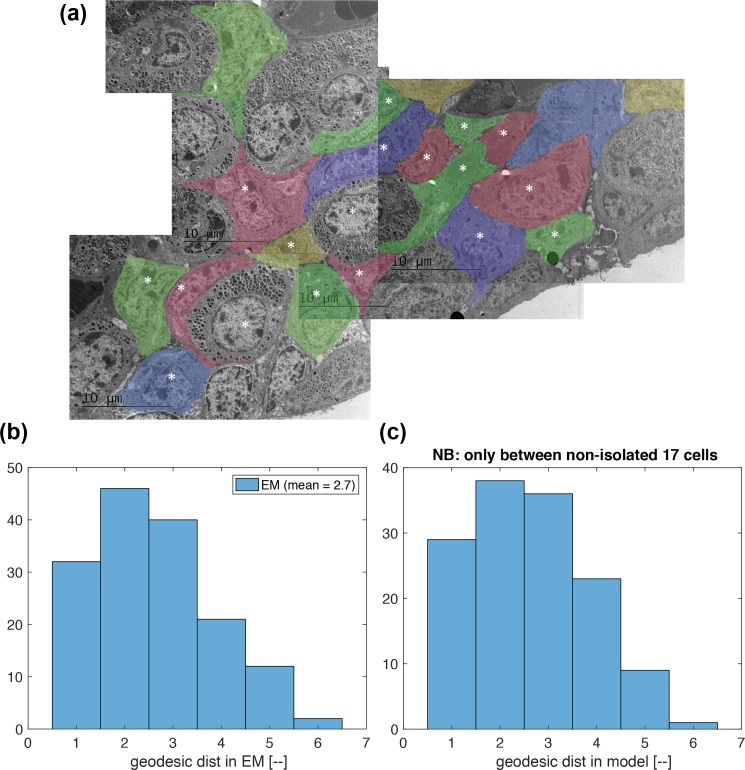
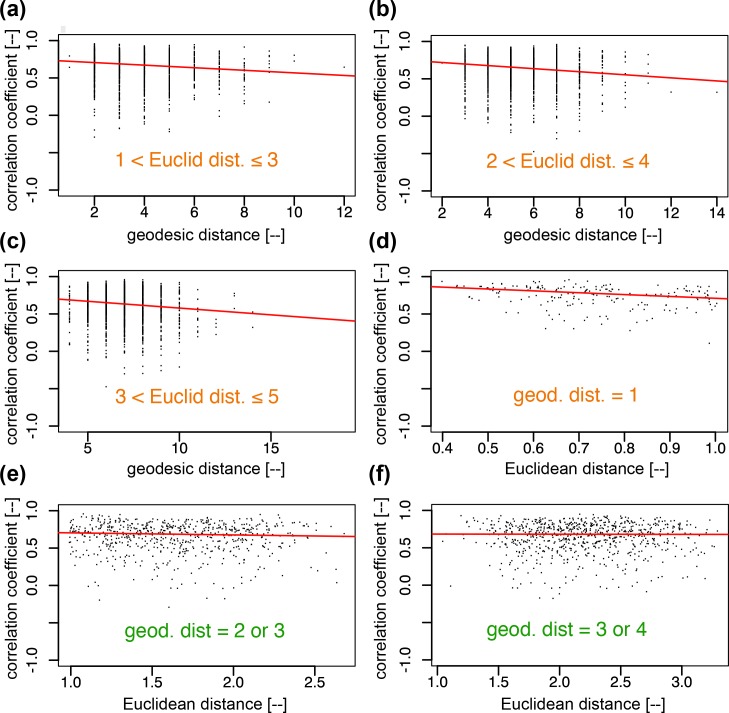
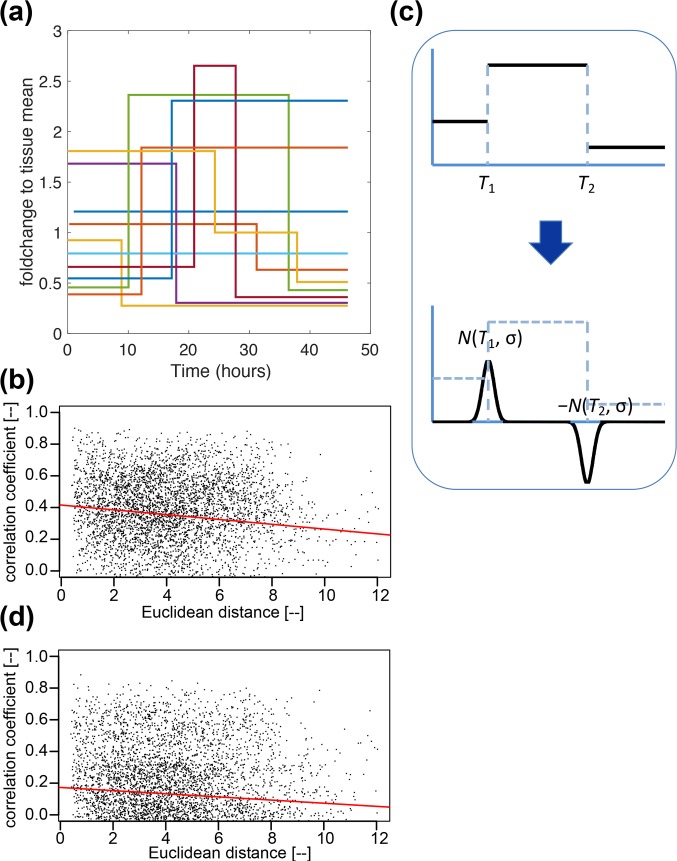
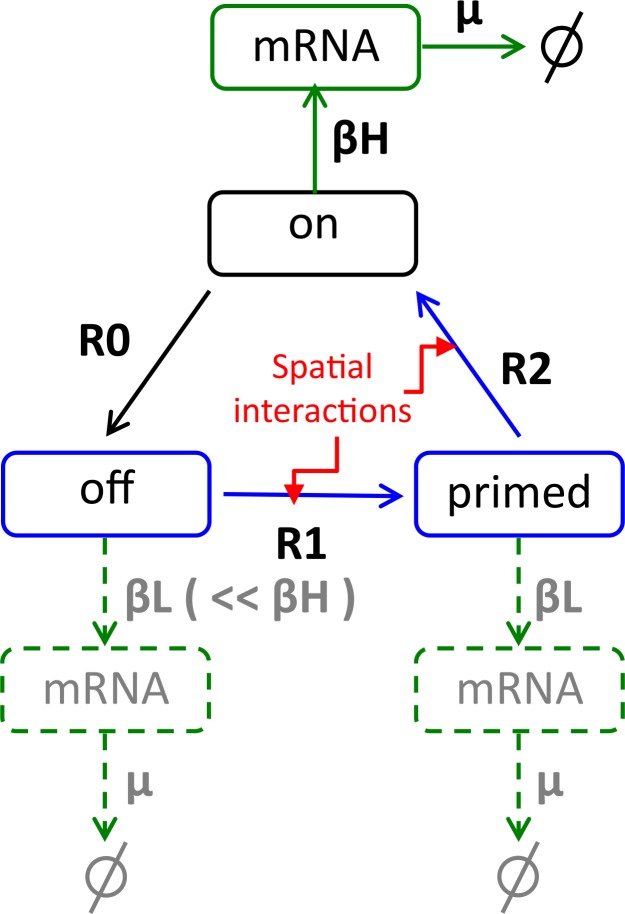
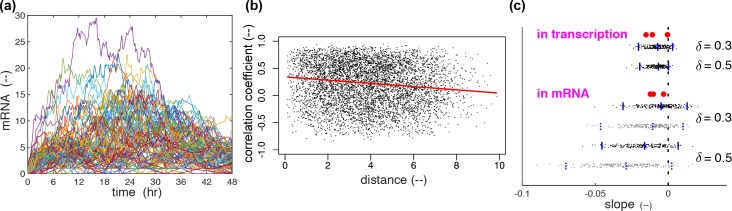
Prolactin is a major hormone product of the pituitary gland, the central endocrine regulator. Despite its physiological importance, the cell-level mechanisms of prolactin production are not well understood. Having significantly improved the resolution of real-time-single-cell-GFP-imaging, the authors recently revealed that prolactin gene transcription is highly dynamic and stochastic yet shows space-time coordination in an intact tissue slice. However, it still remains an open question as to what kind of cellular communication mediates the observed space-time organization. To determine the type of interaction between cells we developed a statistical model. The degree of similarity between two expression time series was studied in terms of two distance measures, Euclidean and geodesic, the latter being a network-theoretic distance defined to be the minimal number of edges between nodes, and this was used to discriminate between juxtacrine from paracrine signalling. The analysis presented here suggests that juxtacrine signalling dominates. To further determine whether the coupling is coordinating transcription or post-transcriptional activities we used stochastic switch modelling to infer the transcriptional profiles of cells and estimated their similarity measures to deduce that their spatial cellular coordination involves coupling of transcription via juxtacrine signalling. We developed a computational model that involves an inter-cell juxtacrine coupling, yielding simulation results that show space-time coordination in the transcription level that is in agreement with the above analysis. The developed model is expected to serve as the prototype for the further study of tissue-level organised gene expression for epigenetically regulated genes, such as prolactin.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Neuregulin-1 (Nrg1) is mainly expressed in rat pituitary gonadotroph cells and possibly regulates prolactin (PRL) secretion in a juxtacrine manner.J Neuroendocrinol. 2011 Dec;23(12):1252-62. doi: 10.1111/j.1365-2826.2011.02223.x. J Neuroendocrinol. 2011. PMID: 21919974
-
Dynamic organisation of prolactin gene expression in living pituitary tissue.J Cell Sci. 2010 Feb 1;123(Pt 3):424-30. doi: 10.1242/jcs.060434. J Cell Sci. 2010. PMID: 20130141 Free PMC article.
-
Autocrine/paracrine roles of extrapituitary growth hormone and prolactin in health and disease: An overview.Gen Comp Endocrinol. 2015 Sep 1;220:103-11. doi: 10.1016/j.ygcen.2014.11.004. Epub 2014 Nov 15. Gen Comp Endocrinol. 2015. PMID: 25448258 Review.
-
Sphingosine 1-phosphate, a key mediator of the cytokine network: juxtacrine signaling.Cytokine Growth Factor Rev. 2011 Feb;22(1):45-53. doi: 10.1016/j.cytogfr.2010.09.004. Epub 2010 Nov 3. Cytokine Growth Factor Rev. 2011. PMID: 21051273 Review.
-
Dynamic analysis of stochastic transcription cycles.PLoS Biol. 2011 Apr;9(4):e1000607. doi: 10.1371/journal.pbio.1000607. Epub 2011 Apr 12. PLoS Biol. 2011. PMID: 21532732 Free PMC article.
Cited by
-
Robust mapping of spatiotemporal trajectories and cell-cell interactions in healthy and diseased tissues.Nat Commun. 2023 Nov 25;14(1):7739. doi: 10.1038/s41467-023-43120-6. Nat Commun. 2023. PMID: 38007580 Free PMC article.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
