

Profiling the genome-wide landscape of tandem repeat expansions
- PMID: 31194863
- PMCID: PMC6735967
- DOI: 10.1093/nar/gkz501
Profiling the genome-wide landscape of tandem repeat expansions
Abstract
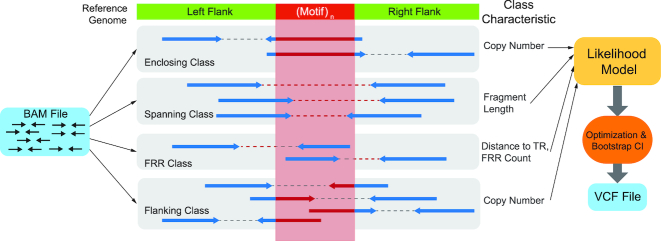
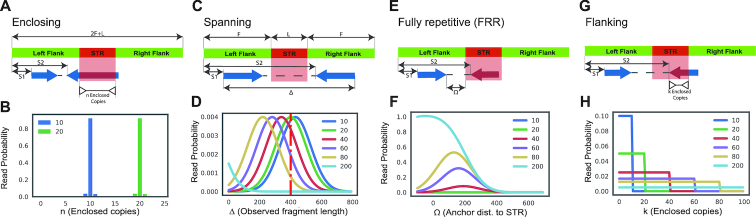
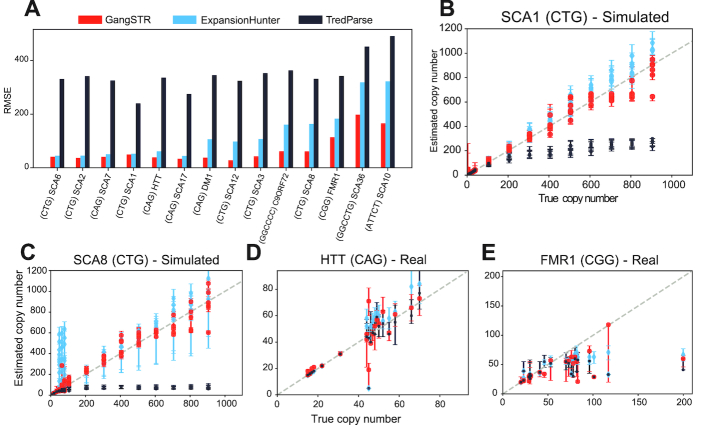
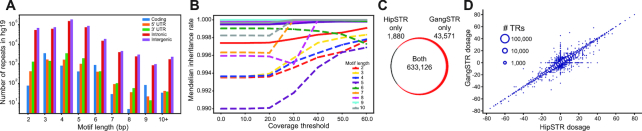
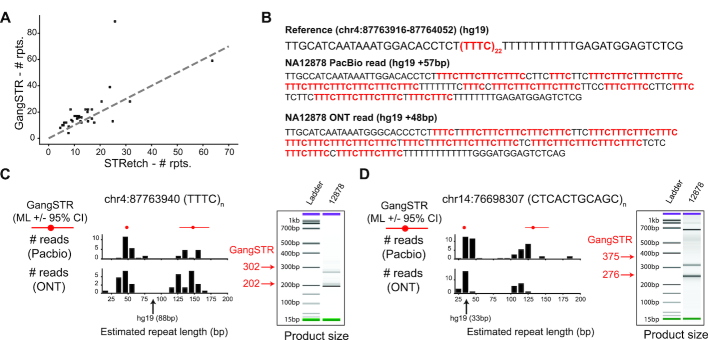
Tandem repeat (TR) expansions have been implicated in dozens of genetic diseases, including Huntington's Disease, Fragile X Syndrome, and hereditary ataxias. Furthermore, TRs have recently been implicated in a range of complex traits, including gene expression and cancer risk. While the human genome harbors hundreds of thousands of TRs, analysis of TR expansions has been mainly limited to known pathogenic loci. A major challenge is that expanded repeats are beyond the read length of most next-generation sequencing (NGS) datasets and are not profiled by existing genome-wide tools. We present GangSTR, a novel algorithm for genome-wide genotyping of both short and expanded TRs. GangSTR extracts information from paired-end reads into a unified model to estimate maximum likelihood TR lengths. We validate GangSTR on real and simulated data and show that GangSTR outperforms alternative methods in both accuracy and speed. We apply GangSTR to a deeply sequenced trio to profile the landscape of TR expansions in a healthy family and validate novel expansions using orthogonal technologies. Our analysis reveals that healthy individuals harbor dozens of long TR alleles not captured by current genome-wide methods. GangSTR will likely enable discovery of novel disease-associated variants not currently accessible from NGS.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Wrzeszczynski K.O., Felice V., Shah M., Rahman S., Emde A.K., Jobanputra V., Frank M.O., Darnell R.B.. Whole genome sequencing-based discovery of structural variants in glioblastoma. Methods Mol. Biol. 2018; 1741:1–29. - PubMed
