

Heart failure drug proscillaridin A targets MYC overexpressing leukemia through global loss of lysine acetylation
- PMID: 31196146
- PMCID: PMC6563382
- DOI: 10.1186/s13046-019-1242-8
Heart failure drug proscillaridin A targets MYC overexpressing leukemia through global loss of lysine acetylation
Abstract
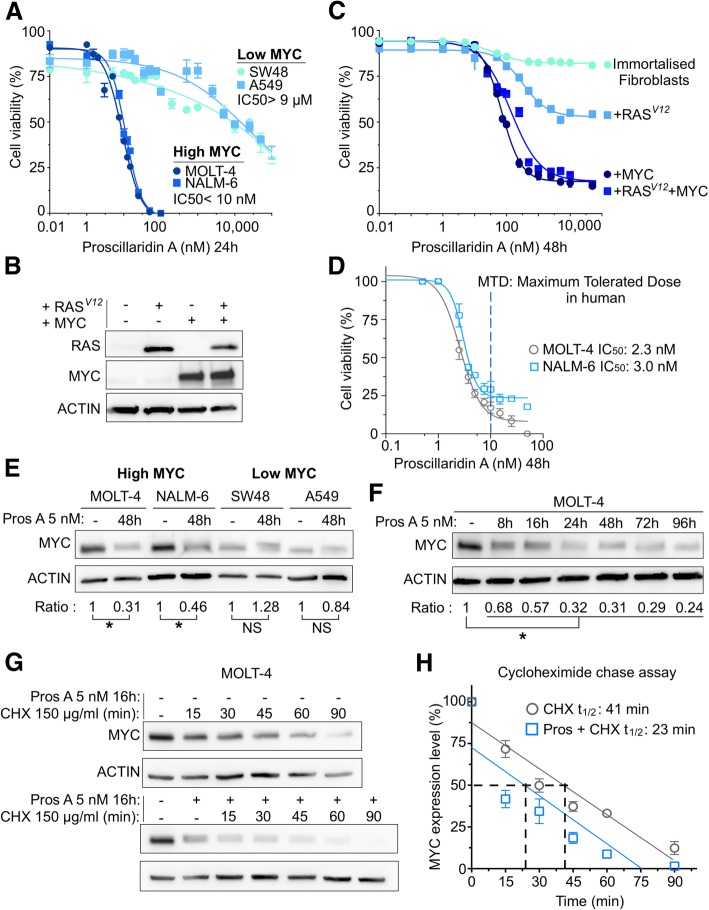
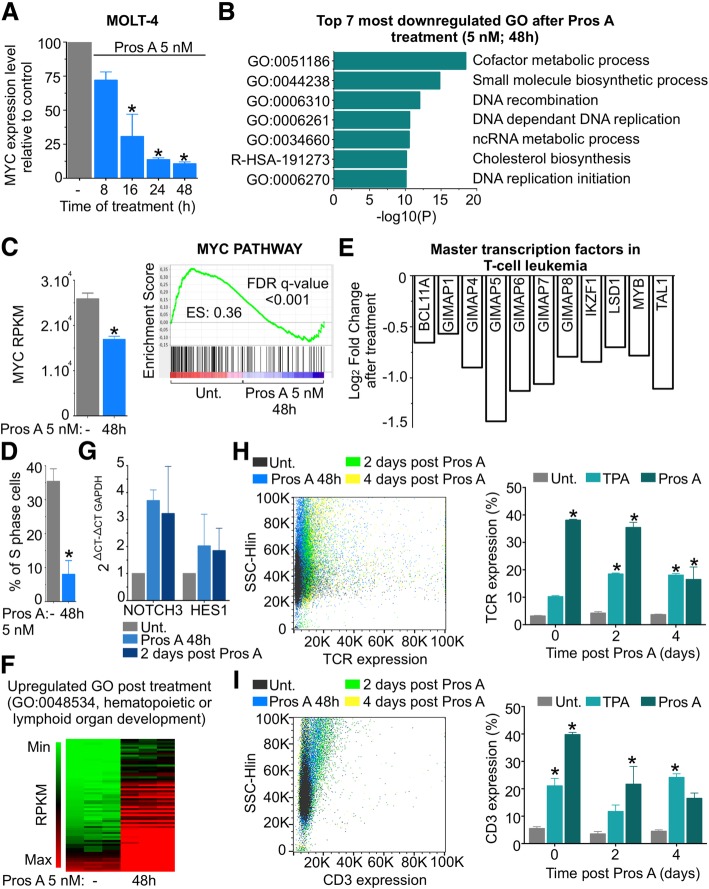
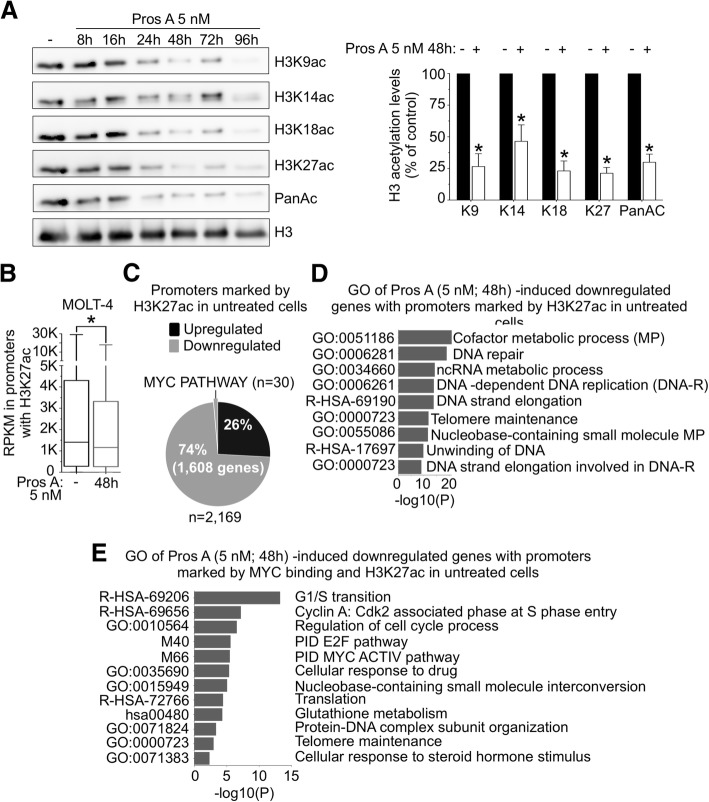
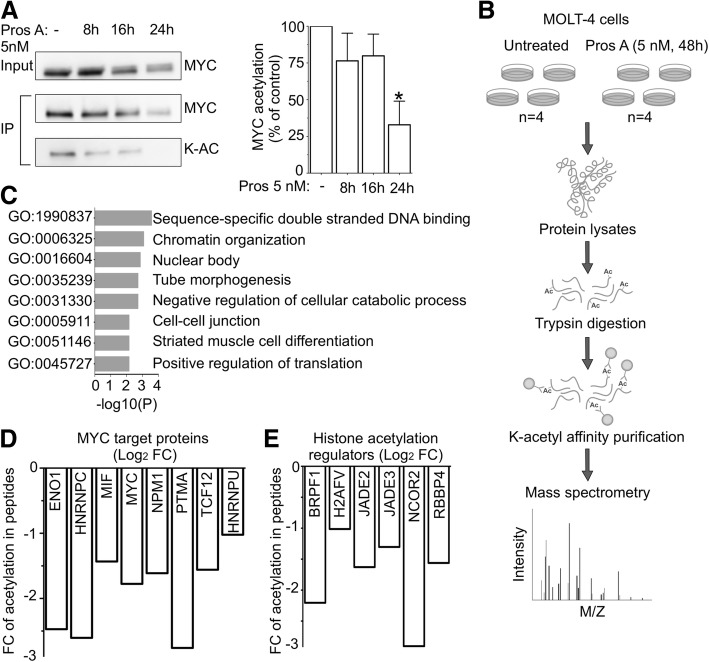
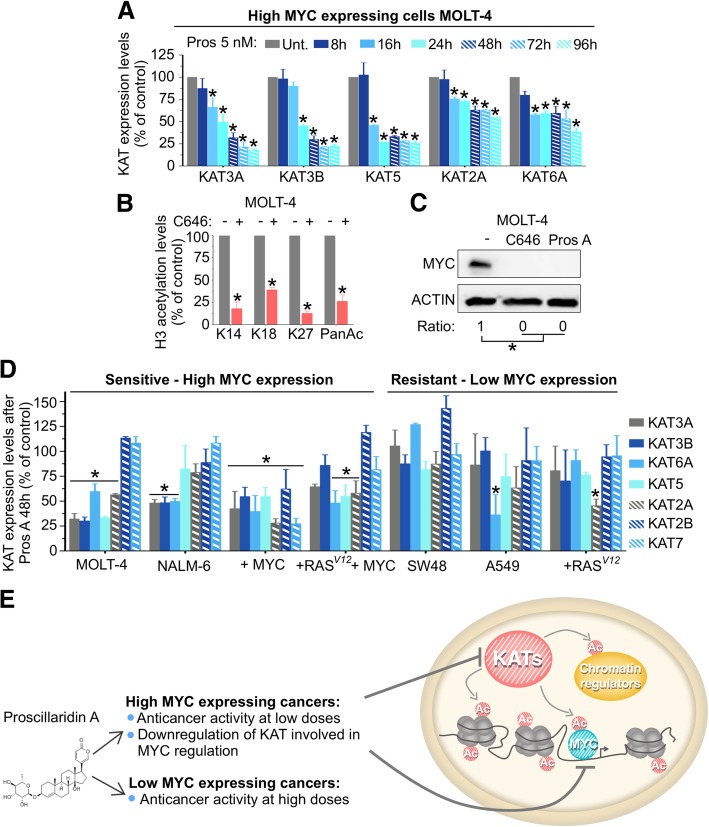
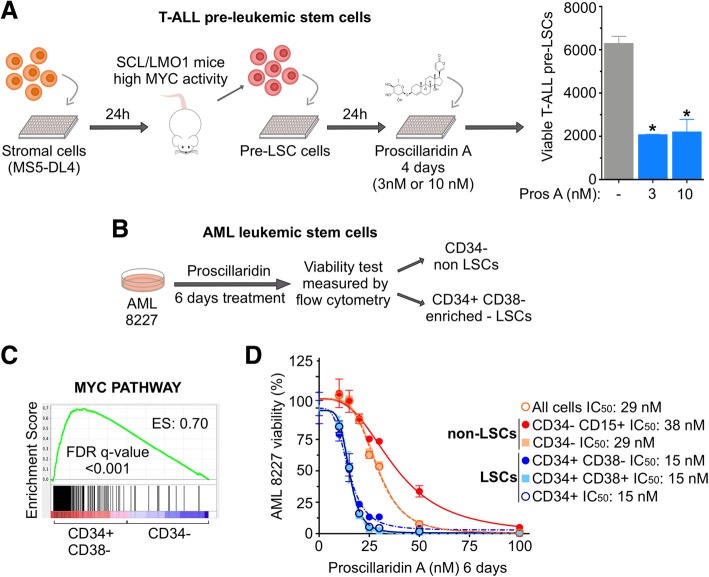
Background: Cardiac glycosides are approved for the treatment of heart failure as Na+/K+ pump inhibitors. Their repurposing in oncology is currently investigated in preclinical and clinical studies. However, the identification of a specific cancer type defined by a molecular signature to design targeted clinical trials with cardiac glycosides remains to be characterized. Here, we demonstrate that cardiac glycoside proscillaridin A specifically targets MYC overexpressing leukemia cells and leukemia stem cells by causing MYC degradation, epigenetic reprogramming and leukemia differentiation through loss of lysine acetylation.
Methods: Proscillaridin A anticancer activity was investigated against a panel of human leukemia and solid tumor cell lines with different MYC expression levels, overexpression in vitro systems and leukemia stem cells. RNA-sequencing and differentiation studies were used to characterize transcriptional and phenotypic changes. Drug-induced epigenetic changes were studied by chromatin post-translational modification analysis, expression of chromatin regulators, chromatin immunoprecipitation, and mass-spectrometry.
Results: At a clinically relevant dose, proscillaridin A rapidly altered MYC protein half-life causing MYC degradation and growth inhibition. Transcriptomic profile of leukemic cells after treatment showed a downregulation of genes involved in MYC pathways, cell replication and an upregulation of hematopoietic differentiation genes. Functional studies confirmed cell cycle inhibition and the onset of leukemia differentiation even after drug removal. Proscillaridin A induced a significant loss of lysine acetylation in histone H3 (at lysine 9, 14, 18 and 27) and in non-histone proteins such as MYC itself, MYC target proteins, and a series of histone acetylation regulators. Global loss of acetylation correlated with the rapid downregulation of histone acetyltransferases. Importantly, proscillaridin A demonstrated anticancer activity against lymphoid and myeloid stem cell populations characterized by MYC overexpression.
Conclusion: Overall, these results strongly support the repurposing of proscillaridin A in MYC overexpressing leukemia.
Keywords: Cardiac glycosides; Chromatin remodelling; Leukemia; Leukemia stem cells; Lysine acetylation; Lysine acetyltransferase; MYC; Proscillaridin A.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Repurposing proscillaridin A in combination with decitabine against embryonal rhabdomyosarcoma RD cells.Cancer Chemother Pharmacol. 2021 Nov;88(5):845-856. doi: 10.1007/s00280-021-04339-6. Epub 2021 Jul 31. Cancer Chemother Pharmacol. 2021. PMID: 34331108
-
Analysis of Myc-induced histone modifications on target chromatin.PLoS One. 2008;3(11):e3650. doi: 10.1371/journal.pone.0003650. Epub 2008 Nov 5. PLoS One. 2008. PMID: 18985155 Free PMC article.
-
High histone acetylation and decreased polycomb repressive complex 2 member levels regulate gene specific transcriptional changes during early embryonic stem cell differentiation induced by retinoic acid.Stem Cells. 2007 Sep;25(9):2191-9. doi: 10.1634/stemcells.2007-0203. Epub 2007 May 24. Stem Cells. 2007. PMID: 17525233
-
Histone deacetylase inhibitors: molecular and biological activity as a premise to clinical application.Curr Drug Metab. 2007 May;8(4):383-93. doi: 10.2174/138920007780655397. Curr Drug Metab. 2007. PMID: 17504226 Review.
-
The key roles of the lysine acetyltransferases KAT6A and KAT6B in physiology and pathology.Drug Resist Updat. 2020 Dec;53:100729. doi: 10.1016/j.drup.2020.100729. Epub 2020 Oct 7. Drug Resist Updat. 2020. PMID: 33130515 Review.
Cited by
-
Cardiac glycoside ouabain efficiently targets leukemic stem cell apoptotic machinery independent of cell differentiation status.Cell Commun Signal. 2023 Oct 12;21(1):283. doi: 10.1186/s12964-023-01317-8. Cell Commun Signal. 2023. PMID: 37828578 Free PMC article.
-
The Marine Natural Compound Dragmacidin D Selectively Induces Apoptosis in Triple-Negative Breast Cancer Spheroids.Mar Drugs. 2023 Dec 15;21(12):642. doi: 10.3390/md21120642. Mar Drugs. 2023. PMID: 38132962 Free PMC article.
-
Anticancer and Antiviral Properties of Cardiac Glycosides: A Review to Explore the Mechanism of Actions.Molecules. 2020 Aug 7;25(16):3596. doi: 10.3390/molecules25163596. Molecules. 2020. PMID: 32784680 Free PMC article. Review.
-
Triple-Negative Breast Cancer Cells Exhibit Differential Sensitivity to Cardenolides from Calotropis gigantea.J Nat Prod. 2020 Jul 24;83(7):2269-2280. doi: 10.1021/acs.jnatprod.0c00423. Epub 2020 Jul 10. J Nat Prod. 2020. PMID: 32649211 Free PMC article.
-
Proscillaridin A Sensitizes Human Colon Cancer Cells to TRAIL-Induced Cell Death.Int J Mol Sci. 2022 Jun 23;23(13):6973. doi: 10.3390/ijms23136973. Int J Mol Sci. 2022. PMID: 35805980 Free PMC article.
References
-
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199–207. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
